| Parikh–Doering oxidation | |
|---|---|
| Named after | Jekishan R. Parikh William von Eggers Doering |
| Reaction type | Organic redox reaction |
The Parikh–Doering oxidation is an oxidation reaction that transforms primary and secondary alcohols into aldehydes and ketones, respectively.[1] The procedure uses dimethyl sulfoxide (DMSO) as the oxidant and the solvent, activated by the sulfur trioxide pyridine complex (SO3•C5H5N) in the presence of triethylamine or diisopropylethylamine as base. Dichloromethane is frequently used as a cosolvent for the reaction.
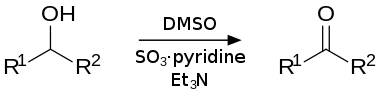
Compared to other activated DMSO oxidations, the Parikh–Doering oxidation is operationally simple: the reaction can be run at non-cryogenic temperatures, often between 0 °C and room temperature, without formation of significant amounts of methyl thiomethylether side products.[2] However, the Parikh–Doering oxidation sometimes requires a large excess of DMSO, SO3•C5H5N and/or base as well as prolonged reaction times for high conversions and yields to be obtained. The following example from the total synthesis of (–)-kumausallene by P.A. Evans and coworkers illustrates typical reaction conditions:[3]
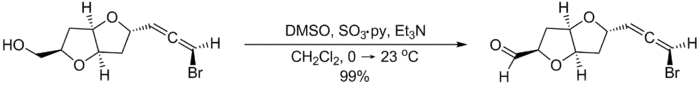
Mechanism
The first step of the Parikh–Doering oxidation is the reaction of dimethyl sulfoxide (DMSO), which exists as a hybrid of the resonance structures 1a and 1b, with sulfur trioxide (2), giving intermediate 3. Nucleophilic attack by alcohol 4 and deprotonation by pyridine (5) gives intermediate 6, an alkoxysulfonium ion associated with the anionic pyridinium sulfate complex.

The addition of at least two equivalents of base deprotonates the alkoxysulfonium ion to give sulfur ylide 7 and removes the pyridinium sulfate counterion. In the last step, the ylide goes through a five-membered ring transition state to give the desired ketone or aldehyde 8, as well as an equivalent of dimethyl sulfide.
.png.webp)
Application
Parikh–Doering oxidation is widely applied in organic synthesis. Here is an example of the Parikh–Doering oxidation's application in the Nicolaou cortistatin total synthesis,[4] where the reaction transforms the hydroxyl functional group into an aldehyde. This process leads to Ohira-Bestmann homologation, which is critical in the following 1,4 addition/aldol condensation/dehydration cascade that forms cortistatins' seven-membered ring. The synthetic route is shown below:

See also
References
- ↑ Tidwell, Thomas T. (1990). "Oxidation of Alcohols to Carbonyl Compounds via Alkoxysulfonium Ylides: The Moffatt, Swern, and Related Oxidations". Organic Reactions. 39: 297. doi:10.1002/0471264180.or039.03. ISBN 0471264180.
- ↑ Parikh, J. R.; Doering, W. v. E. (1967). "Sulfur Trioxide in the Oxidation of Alcohols by Dimethyl Sulfoxide". Journal of the American Chemical Society. 89 (21): 5505–5507. doi:10.1021/ja00997a067.
- ↑ Evans, P. A.; Murthy, V. S.; Roseman, J. D.; Rheingold, A. L. (1999). "Enantioselective Total Synthesis of the Nonisoprenoid Sesquiterpene (-)-Kumausallene". Angewandte Chemie International Edition. 38 (21): 3175–3177. doi:10.1002/(SICI)1521-3773(19991102)38:21<3175::AID-ANIE3175>3.0.CO;2-M. PMID 10556893.
- ↑ Nicolaou, K. C.; Peng, Xiao-Shui; Sun, Ya-Ping; Polet, Damien; Zou, Bin; Lim, Chek Shik; Chen, David Y.-K. (2009). "Total Synthesis and Biological Evaluation of Cortistatins A and J and Analogues Thereof". Journal of the American Chemical Society. 131 (30): 10587–10597. doi:10.1021/ja902939t. PMID 19722632.