Phenol sulfur transferase deficiency, in short PST deficiency, is the lack or the reduced activity of the functional enzyme phenol sulfur transferase, which is crucial in the detoxification of mainly phenolic compounds by catalysing the sulfate conjugation of the hydroxyl groups in the toxic phenolic compounds to result in more hydrophilic forms for more efficient excretion. This metabolic disorder was first discovered in the late 1990s by Dr. Rosemary Waring during her researches with autistic children, which also made this deficiency commonly associated to the topics of autism. Mutations in the PST genes account for the genetic causes of the deficiency, of which single nucleotide polymorphism and methylation of promoters are two examples of mutations that respectively cause conformational abnormalities and diminished expressions to the enzyme, resulting in the reduced detoxification of phenolic compounds and regulation of phenolic neurotransmitter. The deficiency may cause symptoms like flushing, tachycardia, and depression, and be a risk factor for disorders like autism, migraine, and cancer, while it also limits the use of phenolic drugs in PST deficient patients. There is currently no drug available for treating PST deficiency. However, some people suffering from PST deficiency have found taking a digestive enzyme supplement containing Xylanase 10 minutes before eating to greatly reduce symptoms.
Phenol sulfur transferase
Phenol sulfur transferase, in short PST or SULT1, is a subfamily of the enzyme cytosolic sulfotransferases (SULTs) consisting of at least 8 isoforms in humans[1] that catalyze the transfer of sulfuryl group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to phenolic compounds,[2] resulting in more hydrophilic products that can be more easily expelled from tissues for excretion.[3] At high concentration, PST could also catalyze the sulfate conjugation of amino groups.[4] This enzyme subfamily, which exists in nearly all human tissues,[5] is important for the detoxification of phenol-containing xenobiotics or endogenous compounds,[6] including the biotransformation of neurotransmitters and drugs.[5] Its expression is controlled by the PST genes located on chromosomes 2, 4, and 16 depending on the isoform,[7] for example the genes for the predominant isoform throughout the body of human adults, SULT1A1,[8][9] which is highly heritable and variable between individuals,[10] and the most important one in the nervous system, SULT1A3,[11] are located on chromosome 16 at the position of 16p11.2 to 16p12.1.[12]
Discovery
PST deficiency was first discovered in the late 1990s by Dr. Rosemary Waring through a series of tests during her researches on the mechanisms and characteristics of sulfation in autistic children.[13] From the result of the test administering individuals with paracetamol, it was found that the level of sulfate conjugate in urine was significantly lower in the autistic individuals as compared to the non-autistic controls, which was caused by the decreased ability in the formation of sulfated metabolites.[14] The level of sulfate in plasma was also found to be significantly lower in autistic children, leading to a reduced activity of PST.[15] Therefore, she concluded that there was possibly a deficiency of PST in autistic children due to the reduction of sulfate in plasma as a substrate of PST.[13]
Pathophysiology
Causes
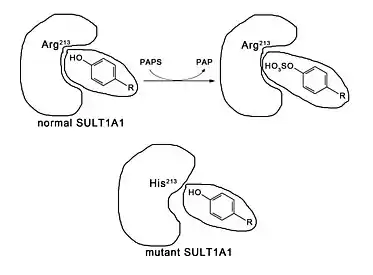
PST deficiency can be caused by inherited mutations in the PST genes,[10] for example the SULT1A1*2 polymorphism, which is a single nucleotide polymorphism at the 638th base of the SULT1A1 gene from guanine to adenosine that causes the change of the 213th amino acid residue of the resultant SULT1A1 from arginine to histidine.[17][18] This mutation causes a conformational change in the enzyme, reducing the size of the binding site and altering the thermochemical properties, which halves the substrate binding affinity and enzyme thermostability,[19] and results in diminished enzymatic activity.[16]
The methylation at the distal and proximal promoters of the PST gens is another mutation that accounts for the deficiency, which causes a reduction in PST expression rather than conformational abnormalities. This prevents the binding of RNA polymerase, which therefore inhibits the mRNA expression of the gene for the production of PST, and finally results in PST deficiency.[20]
Disease-causing mechanisms
PST deficiency can directly cause diseases by the resulted phenol sulfoconjugation defect which reduces the removal of toxic phenolic compounds.[21]
In the liver, where PST serves as one of the important enzymes involved in detoxification, the reduced transcriptional and translational levels of the PST genes would lead to the accumulation of phenolic xenobiotics and cause liver diseases like hepatic steatosis and cirrhosis,[22] or even liver cancers like hepatocellular carcinoma when phenolic carcinogens are accumulated to trigger their developments.[23]
In clinical neurochemistry, PST, in particular the SULT1A3 isoform, is responsible for the degradation of phenolic neurotransmitters such as dopamine and norepinephrine, and therefore is important in the regulation of neurotransmitters which would greatly affect neurological functions. Deficiency or down-regulation of SULT1A3 would cause the retention of neurotransmitter in synapses which affects brain functions including cognitive flexibility and associative learning.[11][24]
Clinical impact
Related disorders
Symptoms of PST deficiency are mainly resulted from the disruptions in multiple metabolic processes due to the accumulation of phenols in the body. Common symptoms include polydipsia, flushing, tachycardia, night sweats, and gastrointestinal problems such as diarrhoea.[13] Neurological and psychiatric disorders such as depression may also occur when regulation of phenolic neurotransmitters is disrupted.[25] PST deficiency is also a risk factor for various diseases including autism, migraine, and cancers.
Autism
It is suspected that mutations, including both microdeletion and microduplication, of the PST genes are the risk factors of autism spectrum disorder,[26] especially the mutation causing decreased SULT1A activity which is usually reported in autistic individuals.[1] Some studies have found that sulfotransferases like PST are involved in glycosylation, and therefore PST deficiency may cause impaired glycosylation, leading to dystroglycanopathies where severe abnormalities of the central nervous system including neuronal migration and cortical defects would occur, and finally result in autistic behaviours.[27] However, it is still unclear on whether PST deficiency is a cause of autism, or just a biomarker for the disorder.[28] Although recent researches have associated autism with the mutations in the position 16p11.2 on chromosome 16,[29][30] where the gene of the predominant PST isoform in the nervous system SULT1A3 exists,[11] due to the large number of gene in this region, PST deficiency resulted from the mutation there may not be a cause of autism but just a condition that is associated with the mutation of another gene which is causing autism.[31][28]
Migraine
PST deficiency in platelets is a risk factor of migraine.[32] It is believed that the reduced PST levels and activity raise the amount of unconjugated amines in the bloodstream and the central nervous system, resulting in a rise of catecholamine level which contributes to the occurrence of recurring headache in migraine.[33] It is also found that dietary intake of foods that are rich in amines may further lower the activity of PST and trigger more serious migraine symptoms.[33]
Cancers
It is controversial for whether PST deficiency increases or decreases the risk of cancers.[34] Although one major function of PST is to inactivate phenolic carcinogens, and therefore a deficiency of PST would reduce inactivation of carcinogens and result in a higher risk of cancer, some studies have also found that PST, specifically SULT1A1, is responsible for the toxification of dietary and environmental mutagens which would increase the risk of cancer, and therefore a decreased risk may be associated with the deficient state of SULT1A1.[19]
Pharmacological impacts
Drug metabolism of phenolic drugs, such as paracetamol and salicylamide, is greatly dependent on the phenol sulfoconjugation by PST, and therefore careful controls on the dosage forms, routes, rates, and duration of administration of those drugs are important for PST deficient patients to prevent accumulation of drugs in the body and depletion of PST for the sulfoconjugation of other xenobiotics and endogenous substances.[35] High dosage of nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin, would also cause a short term inhibition to the activity of PST, and should be administered to PST deficient patients with caution to prevent further reduction in PST activity and accumulation of phenolic compounds which would result in adverse impacts.[14]
References
- 1 2 Salman ED, Kadlubar SA, Falany CN (April 2009). "Expression and localization of cytosolic sulfotransferase (SULT) 1A1 and SULT1A3 in normal human brain". Drug Metabolism and Disposition. 37 (4): 706–9. doi:10.1124/dmd.108.025767. PMC 2680540. PMID 19171676.
- ↑ Duffel MW, Marshal AD, McPhie P, Sharma V, Jakoby WB (2001). "Enzymatic aspects of the phenol (aryl) sulfotransferases". Drug Metabolism Reviews. 33 (3–4): 369–95. doi:10.1081/dmr-120001394. PMID 11768773. S2CID 1975310.
- ↑ Dooley TP (February 1998). "Cloning of the human phenol sulfotransferase gene family: three genes implicated in the metabolism of catecholamines, thyroid hormones and drugs". Chemico-Biological Interactions. 109 (1–3): 29–41. doi:10.1016/S0009-2797(97)00118-X. PMID 9566731.
- ↑ Coughtrie MW (July 1996). "Sulphation catalysed by the human cytosolic sulphotransferases--chemical defence or molecular terrorism?". Human & Experimental Toxicology. 15 (7): 547–55. doi:10.1177/096032719601500701. PMID 8818707. S2CID 10698535.
- 1 2 Weinshilboum R (March 1988). "Phenol sulfotransferase inheritance". Cellular and Molecular Neurobiology. 8 (1): 27–34. doi:10.1007/bf00712908. PMID 3042142. S2CID 27453493.
- ↑ Sekura RD, Jakoby WB (July 1979). "Phenol sulfotransferases". The Journal of Biological Chemistry. 254 (13): 5658–63. doi:10.1016/S0021-9258(18)50465-8. PMID 447677.
- ↑ Gamage N, Barnett A, Hempel N, Duggleby RG, Windmill KF, Martin JL, McManus ME (March 2006). "Human sulfotransferases and their role in chemical metabolism". Toxicological Sciences. 90 (1): 5–22. CiteSeerX 10.1.1.319.6099. doi:10.1093/toxsci/kfj061. PMID 16322073.
- ↑ Chen G, Chen X (September 2003). "Arginine residues in the active site of human phenol sulfotransferase (SULT1A1)". The Journal of Biological Chemistry. 278 (38): 36358–64. doi:10.1074/jbc.M306045200. PMC 3118444. PMID 12867416.
- ↑ Cook I, Wang T, Girvin M, Leyh TS (December 2016). "The structure of the catechin-binding site of human sulfotransferase 1A1". Proceedings of the National Academy of Sciences of the United States of America. 113 (50): 14312–14317. doi:10.1073/pnas.1613913113. PMC 5167148. PMID 27911811.
- 1 2 Yu X, Dhakal IB, Beggs M, Edavana VK, Williams S, Zhang X, et al. (December 2010). "Functional genetic variants in the 3'-untranslated region of sulfotransferase isoform 1A1 (SULT1A1) and their effect on enzymatic activity". Toxicological Sciences. 118 (2): 391–403. doi:10.1093/toxsci/kfq296. PMC 2984532. PMID 20881232.
- 1 2 3 Darrah K, Wang T, Cook I, Cacace M, Deiters A, Leyh TS (February 2019). "Allosteres to regulate neurotransmitter sulfonation". The Journal of Biological Chemistry. 294 (7): 2293–2301. doi:10.1074/jbc.RA118.006511. PMC 6378965. PMID 30545938.
- ↑ Dooley TP, Obermoeller RD, Leiter EH, Chapman HD, Falany CN, Deng Z, Siciliano MJ (November 1993). "Mapping of the phenol sulfotransferase gene (STP) to human chromosome 16p12.1-p11.2 and to mouse chromosome 7". Genomics. 18 (2): 440–3. doi:10.1006/geno.1993.1494. PMID 8288252.
- 1 2 3 O'Reilly BA, Waring RH (1993). "Enzyme and sulphur oxidation deficiencies in autistic children with known food intolerances" (PDF). Journal of Orthomolecular Medicine. 8 (4): 198–200.
- 1 2 Alberti A, Pirrone P, Elia M, Waring RH, Romano C (August 1999). "Sulphation deficit in "low-functioning" autistic children: a pilot study". Biological Psychiatry. 46 (3): 420–4. doi:10.1016/s0006-3223(98)00337-0. PMID 10435209. S2CID 20942689.
- ↑ Waring RH, Ngong JM, Klovrza L, Green S, Sharp H (1997). "Biochemical parameters in autistic children". Dev Brain Dysfunct. 10: 43–47. ISSN 1019-5815.
- 1 2 Dash R, Ali MC, Dash N, Azad MA, Hosen SM, Hannan MA, Moon IS (December 2019). "Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding". International Journal of Molecular Sciences. 20 (24): 6256. doi:10.3390/ijms20246256. PMC 6969939. PMID 31835852.
- ↑ Fernández-Santander A, Novillo A, Gaibar M, Romero-Lorca A, Moral P, Sánchez-Cuenca D, et al. (August 2016). "Cytochrome and sulfotransferase gene variation in north African populations". Pharmacogenomics. 17 (13): 1415–23. doi:10.2217/pgs-2016-0016. PMID 27471773.
- ↑ Liang G, Miao X, Zhou Y, Tan W, Lin D (May 2004). "A functional polymorphism in the SULT1A1 gene (G638A) is associated with risk of lung cancer in relation to tobacco smoking". Carcinogenesis. 25 (5): 773–8. doi:10.1093/carcin/bgh053. PMID 14688021.
- 1 2 Kotnis A, Kannan S, Sarin R, Mulherkar R (October 2008). "Case-control study and meta-analysis of SULT1A1 Arg213His polymorphism for gene, ethnicity and environment interaction for cancer risk". British Journal of Cancer. 99 (8): 1340–7. doi:10.1038/sj.bjc.6604683. PMC 2570530. PMID 18854828.
- ↑ Kwon MS, Kim SJ, Lee SY, Jeong JH, Lee ES, Kang HS (January 2006). "Epigenetic silencing of the sulfotransferase 1A1 gene by hypermethylation in breast tissue". Oncology Reports. 15 (1): 27–32. doi:10.3892/or.15.1.27. PMID 16328031.
- ↑ Kuchel O, Buu NT, Serri O (1982). "Sulfoconjugation of catecholamines, nutrition, and hypertension". Hypertension. 4 (5 Pt 2): III93-8. doi:10.1161/01.HYP.4.5_Pt_2.III93. PMID 7049935.
- ↑ Yalcin EB, More V, Neira KL, Lu ZJ, Cherrington NJ, Slitt AL, King RS (September 2013). "Downregulation of sulfotransferase expression and activity in diseased human livers". Drug Metabolism and Disposition. 41 (9): 1642–50. doi:10.1124/dmd.113.050930. PMC 3876809. PMID 23775849.
- ↑ Yeo M, Na YM, Kim DK, Kim YB, Wang HJ, Lee JA, et al. (January 2010). "The loss of phenol sulfotransferase 1 in hepatocellular carcinogenesis". Proteomics. 10 (2): 266–76. doi:10.1002/pmic.200900721. PMID 19904771. S2CID 206364501.
- ↑ Goldstein DS, Swoboda KJ, Miles JM, Coppack SW, Aneman A, Holmes C, et al. (July 1999). "Sources and physiological significance of plasma dopamine sulfate". The Journal of Clinical Endocrinology and Metabolism. 84 (7): 2523–31. doi:10.1210/jcem.84.7.5864. PMID 10404831.
- ↑ Pennings EJ, Van Kempen GM (October 1983). "Assay of phenol sulphotransferase in human blood". Clinica Chimica Acta; International Journal of Clinical Chemistry. 134 (1–2): 199–206. doi:10.1016/0009-8981(83)90197-3. PMID 6580977.
- ↑ Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. (February 2008). "Association between microdeletion and microduplication at 16p11.2 and autism". The New England Journal of Medicine. 358 (7): 667–75. doi:10.1056/NEJMoa075974. PMID 18184952.
- ↑ Dwyer CA, Esko JD (October 2016). "Glycan susceptibility factors in autism spectrum disorders". Molecular Aspects of Medicine. 51: 104–14. doi:10.1016/j.mam.2016.07.001. PMC 5556687. PMID 27418189.
- 1 2 Bjørklund G, Meguid NA, El-Ansary A, El-Bana MA, Dadar M, Aaseth J, et al. (December 2018). "Diagnostic and Severity-Tracking Biomarkers for Autism Spectrum Disorder". Journal of Molecular Neuroscience. 66 (4): 492–511. doi:10.1007/s12031-018-1192-1. PMID 30357679. S2CID 53027975.
- ↑ Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, et al. (November 2016). "16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort". American Journal of Medical Genetics. Part A. 170 (11): 2943–2955. doi:10.1002/ajmg.a.37820. PMID 27410714. S2CID 2469192.
- ↑ Angelakos CC, Watson AJ, O'Brien WT, Krainock KS, Nickl-Jockschat T, Abel T (April 2017). "Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism". Autism Research. 10 (4): 572–584. doi:10.1002/aur.1707. PMC 6201314. PMID 27739237.
- ↑ Arbogast T, Ouagazzal AM, Chevalier C, Kopanitsa M, Afinowi N, Migliavacca E, et al. (February 2016). Barsh GS (ed.). "Reciprocal Effects on Neurocognitive and Metabolic Phenotypes in Mouse Models of 16p11.2 Deletion and Duplication Syndromes". PLOS Genetics. 12 (2): e1005709. doi:10.1371/journal.pgen.1005709. PMC 4752317. PMID 26872257.
- ↑ Littlewood J, Glover V, Sandler M, Petty R, Peatfield R, Rose FC (May 1982). "Platelet phenolsulphotransferase deficiency in dietary migraine". Lancet. 1 (8279): 983–6. doi:10.1016/S0140-6736(82)91990-0. PMID 6122845. S2CID 39305175.
- 1 2 Alam Z, Coombes N, Waring RH, Williams AC, Steventon GB (November 1997). "Platelet sulphotransferase activity, plasma sulphate levels and sulphation capacity in patients with migraine and tension headache". Cephalalgia. 17 (7): 761–4. doi:10.1046/j.1468-2982.1997.1707761.x. PMID 9399006. S2CID 20415750.
- ↑ Liu J, Zhao R, Ye Z, Frey AJ, Schriver ER, Snyder NW, Hebbring SJ (November 2017). "Relationship of SULT1A1 copy number variation with estrogen metabolism and human health". The Journal of Steroid Biochemistry and Molecular Biology. 174: 169–175. doi:10.1016/j.jsbmb.2017.08.017. PMC 5675753. PMID 28867356.
- ↑ Levy G (July 1986). "Sulfate conjugation in drug metabolism: role of inorganic sulfate". Federation Proceedings. 45 (8): 2235–40. PMID 3459670.