| Developer(s) | McGill University McGill Centre for Bioinformatics |
|---|---|
| Initial release | 2010 |
| Engine |
|
| Platform | Unity (web browser, Android, iOS) |
| Available in | English, French |
| Type | Video game |
| Website | phylo |
Phylo is an experimental video game about multiple sequence alignment optimisation.[1] Developed by the McGill Centre for Bioinformatics, it was originally released as a free Flash game in November 2010. Designed as a game with a purpose, players solve pattern-matching puzzles that represent nucleotide sequences of different phylogenetic taxa to optimize alignments over a computer algorithm. By aligning together each nucleotide sequence, represented as differently coloured blocks, players attempt to create the highest point value score for each set of sequences by matching as many colours as possible and minimizing gaps.
The nucleotide sequences generated by Phylo are obtained from actual sequence data from the UCSC Genome Browser. High-scoring player alignments are collected as data and sent back to the McGill Centre for Bioinformatics to be further evaluated with a stronger scoring algorithm. Those player alignments that score higher than the current computer-generated score will be re-introduced into the global alignment as an optimization.
Background
The goal of multiple sequence alignments in phylogenetics is to determine the most likely nucleotide sequence of each species by comparing the sequences of children species with those of a most recent common ancestor. Producing such an optimal multiple sequence alignment is usually determined with a dynamic programming algorithm that finds the most probable evolutionary outcome by minimizing the number of mutations required. These algorithms generate phylogenetic trees for each nucleotide in a sequence for each species, and determine the genetic sequence for a common ancestor by comparing the trees of the child species. The algorithms then score and sort the completed phylogenetic tree, and the alignment with the maximum parsimony score is determined to be the optimal, and thus most evolutionarily likely, multiple sequence alignment. However, finding such an optimal alignment for a large number of sequences has been determined to be an NP-complete problem.
Phylo uses human-based computation to create an interactive genetic algorithm to solve the multiple sequence alignment problem instead. Generation of the ancestral sequences and parsimony scoring is still computed using a variation of the Fitch–Margoliash method, but Phylo abstracts the genetic sequences obtained from the UCSC Genome Browser into a pattern-matching game, allowing human players to suggest the most likely alignment rather than algorithmically considering all possible trees.
Gameplay
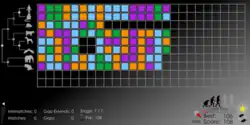
Each puzzle in Phylo is categorized based on the number of total sequence fragments to be aligned and a disease that is associated with that fragment in humans. Once a puzzle is chosen, a few of the genetic sequence fragments for each species to be aligned, represented as coloured blocks, are each placed on a single row of a grid. Each nucleotide of a genetic sequence fragment is free to move along the grid. Players can then adjust the sequences as necessary in order to create the largest number of colour matches in each column between them, while minimizing the number of the gaps that appear.
Scoring of the sequence alignment is done by comparing each of the player-aligned sequences with an algorithm-determined ancestral sequence generated at each node. A colour match yields +1 to the score, a mismatch yields -1, an opening of a gap yields -5, and an extension of any existing gap yields -1. The sum of all comparisons is then determined every several seconds, which provides the final score for that player's alignment. For each puzzle, only a few sequences are initially available at the beginning of the game. A computer-determined par score must be beaten by the player before moving on to the next round and unlocking more sequences to match. A player wins and is allowed to submit their sequence alignment to the database by matching or surpassing the final par score generated by the computer for each puzzle.
Levels
As of May 2019 (v 3.1.5), Phylo comes in three game modes:
- Story mode, with levels arranged in a guided tutorial
- The original Phylo mode, with the choice of diseases
- A new Ribo mode for RNA molecules, where both sequences and RNA secondary structures (stem-loops) are aligned.[2]
Results
Compared to the computer output, players were able to improve 70% of the alignments.[1] In 2013, Phylo developers built a webserver called Open-Phylo (now defunct) that allows researchers to upload their own sets of sequences for players to align. Compared to computer alignments, expert players were able to make mostly small improvements over what sequence alignment algorithms could do. There were also some minor cases of significantly better alignments proposed by humans.[3] An 2017 report on five years of historical Phylo data reaches a similar conclusion.[4]
See also
References
- 1 2 3 Kawrykow, A.; Roumanis, G.; Kam, A.; Kwak, D.; Leung, C.; Wu, C.; Zarour, E.; Phylo, L.; Sarmenta, M.; Blanchette, M.; Waldispühl, J.; Phylo Players (2012). Michalak, Pawel (ed.). "Phylo: A Citizen Science Approach for Improving Multiple Sequence Alignment". PLOS ONE. 7 (3): e31362. Bibcode:2012PLoSO...731362K. doi:10.1371/journal.pone.0031362. PMC 3296692. PMID 22412834.
- ↑ Waldispühl, J; Kam, A; Gardner, PP (2015). "Crowdsourcing RNA structural alignments with an online computer game" (PDF). Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing: 330–41. PMID 25592593.
- ↑ Kwak, D; Kam, A; Becerra, D; Zhou, Q; Hops, A; Zarour, E; Kam, A; Sarmenta, L; Blanchette, M; Waldispühl, J (2013). "Open-Phylo: a customizable crowd-computing platform for multiple sequence alignment". Genome Biology. 14 (10): R116. doi:10.1186/gb-2013-14-10-r116. PMC 4014878. PMID 24148814.
- ↑ Waldispühl, Jérôme; Blanchette, Mathieu; Ahsan, Faizy; Singh, Akash (21 September 2017). "Lessons from an Online Massive Genomics Computer Game". Fifth AAAI Conference on Human Computation and Crowdsourcing.