| Pineoblastoma | |
|---|---|
| Other names | Pineoblastoma |
| Specialty | Oncology |
| Symptoms | Nausea, vomiting, headache, double vision, problems with eye movement[1] |
| Usual onset | Childhood, between the ages of 20 and 40[1] |
| Frequency | Just under half of all pineal gland tumors, which make up fewer than 1% of all primary brain tumors[1] |
Pineoblastoma is a malignant tumor of the pineal gland. A pineoblastoma is a supratentorial midline primitive neuroectodermal tumor.[2] Pineoblastoma can present at any age, but is most common in young children. They account for 0.001% of all primary CNS neoplasms.[3]
Epidemiology
Pineoblastomas typically occur at very young ages. One study found the average age of presentation to be 4.3 years, with peaks at age 3 and 8.[3] Another cites cases to more commonly occur in patients under 2 years of age.[4] Rates of occurrence for males and females are similar, but may be slightly more common in females.[4][3] One study found incidence of pineoblastoma to be increased in black patients compared to white patients by around 71%.[5] This difference was most apparent in patients aged 5 to 9 years old.
Pathophysiology
The pineal gland is a small organ in the center of the brain that is responsible for controlling melatonin secretion.[2] Several tumors can occur in the area of the pineal gland, with the most aggressive being pineoblastoma. Pineoblastomas arise from embryonal cells in the pineal gland and are rapidly growing. They are considered grade 4 tumors, meaning they are malignant and may metastasize.[6] Due to the pineal gland's location at the center of the brain and the rapidly growing nature of this disease, obstruction of CNS fluid is a common symptom.
The exact cause of pineoblastoma is unknown. MicroRNA dysregulation has been found to be associated with many cases of pineoblastoma, specifically, mutations in DICER1 and DROSHA genes.[7] DICER1 germline mutations cause a tumor predisposition syndrome, and should be considered in patients with pineoblastoma.[8]
Pineoblastoma may occur in patients with hereditary uni- or bilateral retinoblastoma. When retinoblastoma patients present with pineoblastoma this is characterized as "trilateral retinoblastoma".[9] Up to 5% of patients with hereditary retinoblastoma are at risk of developing trilateral retinoblastoma.[10] This tumor combination is more aggressive than an isolated pineoblastoma.[4] Prognosis of patients with trilateral retinoblastoma is dismal, only a few patients have survived more than 5 years after diagnosis; all survivors were diagnosed with small tumors in a subclinical stage.[11] Recent advances in (high-dose) chemotherapy treatment regimens and early detection have improved survival of patients with trilateral retinoblastoma to up to 50%.[10]
Additionally, various mutations or deletions in chromosomes 1, 9, 13, 16 and 22 have been associated with pineoblastoma incidence.[3]
Clinical Features
The most common symptoms to occur with pineoblastoma are headache, behavior changes, and cognitive disturbances.[12] These masses also often cause obstructive hydrocephalus, leading to increased intracranial pressure. This can result in vision changes and Parinaud's syndrome.[3]
Due to the aggressive nature of the disease, tumor spread at the time of diagnosis is common.[13] Pineoblastomas often invades locally, with spread to the head and spine seen in 25-41% of patients.[3] While CNS spread is relatively common, these tumors rarely cause distant metastases.[14]
Diagnosis
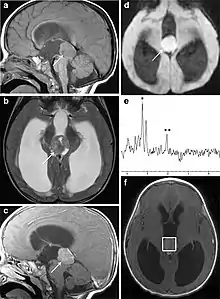
Several imaging methods can be used to diagnose pineoblastoma. Initially, urgent CTs are recommended, followed by MR imaging.[13] CT will show large, multilobulated masses with heterogenous contrast enhancement and peripheral calcification of the pineal gland.[4][3] On MRI, pineoblastomas again appear as masses with heterogenous enhancement. They often appear hypo- to isointense on T1 and slightly hyperintense on T2-weighted images. Some areas of necrosis or hemorrhage may be seen as well. PET-CT has also been used in diagnosis, and shows increased uptake of fludeoxyglucose with pineoblastomas compared to other pineal masses.
Diagnosis also requires CSF sampling via lumbar puncture to assess for cytology and tumor markers.[13]
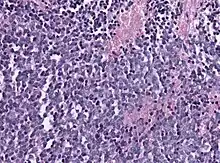
Biopsy is required for diagnosis. Pineoblastomas appear as high grade, highly cellular, small blue cells histologically. Features of aggressive malignancies can be seen, like high nucleus-to-cytoplasm ration, poorly differentiated cells, high mitotic activity, and necrosis.[13][3] Homer Wright, or neuroblastic, and Flexner-Wintersteiner, or retinoblastic, rosettes can also be seen. In contrast to other masses of the pineal gland, pineocytomatous rosettes are not present.[13] Immunohistochemistry staining will reveal neuronal, glial, and photoreceptor marker positivity. This includes synaptophysin, neurofilament protein, and CRX, a specific pineal or retinal marker, positive staining.[13][7]
Treatment
Initial treatment for pineoblastoma often includes a shunting procedure to redirect accumulated cerebrospinal fluid secondary to obstructive hydrocephalus.[2] This shunt can help manage increased intracranial pressure and relieve some symptoms. Surgery to remove the tumor is associated with better outcomes, however, this is not always possible due to the proximity of the pineal gland to neurovascular structures.[3] Complete tumor resection is only seen in about 30% of cases. Following surgery, radiation therapy to the brain and spinal cord can increase survival.[2] However, radiation can only safely be used in patients over 3 years old due to the risk of significant neurological impairment. Chemotherapy treatment can also be used, either before or after surgery; its optimal use is still under investigation.[3]
Prognosis
Pineoblastomas are very aggressive tumors. 5-year survival for patients with pineoblastomas is around 58%.[13] Prognosis for patients under 5 years old is lower, between 15 and 40%.[3] Disseminated disease at diagnosis is also associated with worse outcomes. When pineoblastomas occur with retinoblastomas, the prognosis is typically worse, and these patients require more aggressive treatment.[4]
Complete gross tumor resection is associated with improved prognosis, but is difficult and rare to achieve. Radiation therapy after surgery is also linked to improved survival.[3]
References
- 1 2 3 "Pineoblastoma". St. Jude Children's Research Hospital. Retrieved Mar 8, 2023.
- 1 2 3 4 Mayo Clinic Staff. "Brain Tumor: Pineoblastoma". Retrieved 18 April 2022.
- 1 2 3 4 5 6 7 8 9 10 11 12 Sin-Chan P, Li BK, Ho B, Fonseca A, Huang A (July 2018). "Molecular Classification and Management of Rare Pediatric Embryonal Brain Tumors". Current Oncology Reports. 20 (9): 69. doi:10.1007/s11912-018-0717-7. PMID 29995179.
- 1 2 3 4 5 Tamrazi B, Nelson M, Blüml S (February 2017). "Pineal Region Masses in Pediatric Patients". Neuroimaging Clinics of North America. 27 (1): 85–97. doi:10.1016/j.nic.2016.08.002. PMID 27889025.
- ↑ Greppin K, Cioffi G, Waite KA, Ostrom QT, Landi D, Takaoka K, et al. (April 2022). "Epidemiology of pineoblastoma in the United States, 2000-2017". Neuro-Oncology Practice. 9 (2): 149–157. doi:10.1093/nop/npac009. PMC 8965073. PMID 35371520.
- ↑ "Pineal Region Tumors Diagnosis and Treatment - NCI". www.cancer.gov. 2018-09-17. Retrieved 2022-10-11.
- 1 2 Blessing MM, Alexandrescu S (June 2020). "Embryonal Tumors of the Central Nervous System: An Update". Surgical Pathology Clinics. 13 (2): 235–247. doi:10.1016/j.path.2020.01.003. PMID 32389264.
- ↑ Schultz KA, Stewart DR, Kamihara J, Bauer AJ, Merideth MA, Stratton P, et al. (1993). "DICER1 Tumor Predisposition". In Adam MP, Everman DB, Mirzaa GM, Pagon RA (eds.). GeneReviews®. Seattle (WA): University of Washington, Seattle. PMID 24761742. Retrieved 2022-10-11.
- ↑ Provenzale JM, Weber AL, Klintworth GK, McLendon RE (January 1995). "Radiologic-pathologic correlation. Bilateral retinoblastoma with coexistent pinealoblastoma (trilateral retinoblastoma)". AJNR. American Journal of Neuroradiology. 16 (1): 157–165. PMID 7900586.
- 1 2 de Jong MC, Kors WA, de Graaf P, Castelijns JA, Moll AC, Kivelä T (December 2015). "The Incidence of Trilateral Retinoblastoma: A Systematic Review and Meta-Analysis". American Journal of Ophthalmology. 160 (6): 1116–1126.e5. doi:10.1016/j.ajo.2015.09.009. hdl:10138/223832. PMID 26374932.
- ↑ Kivelä T (June 1999). "Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma". Journal of Clinical Oncology. 17 (6): 1829–1837. doi:10.1200/JCO.1999.17.6.1829. PMID 10561222.
- ↑ "Pineoblastoma - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Retrieved 2022-10-06.
- 1 2 3 4 5 6 7 Amato-Watkins AC, Lammie A, Hayhurst C, Leach P (2016-01-02). "Pineal parenchymal tumours of intermediate differentiation - An evidence-based review of a new pathological entity". British Journal of Neurosurgery. 30 (1): 11–15. doi:10.3109/02688697.2015.1096912. PMID 26571134.
- ↑ "Pineoblastoma". www.stjude.org. Retrieved 2022-10-05.