 | |
| Filename extension |
.json |
|---|---|
| Internet media type |
application/json |
| Developed by | DeepMind, EMBL-EBI |
| Type of format | Bioinformatics |
| Website | https://alphafold.ebi.ac.uk/faq |
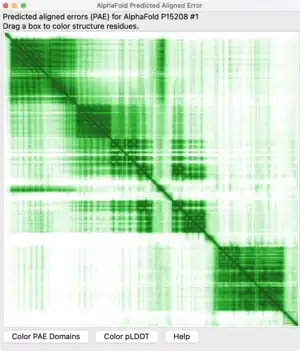
The Predicted Aligned Error (PAE) is a quantitative output produced by AlphaFold, a protein structure prediction system developed by DeepMind.[1] PAE estimates the expected positional error for each residue in a predicted protein structure if it were aligned to a corresponding residue in the true protein structure. This measurement helps scientists assess the confidence in the relative positions and orientations of different parts of the predicted protein model.[2]
Structure
PAE is presented as a two-dimensional (2D) interactive plot where the color at coordinates (x, y) represents the predicted position error at residue x if the predicted and true structures were aligned on residue y.[3] Lower PAE values for residue pairs from different domains suggest well-defined relative positions and orientations in the prediction, while higher PAE values indicate uncertainty in the relative positions or orientations.
Users can download the raw PAE data for all residue pairs in a custom JSON format for further analysis or visualization using a programming language such as Python. The format of the JSON file is as follows:
[
{
"predicted_aligned_error": [[0, 1, 4, 7, 9, ...], ...],
"max_predicted_aligned_error": 31.75
}
]
In the JSON file, the field predicted_aligned_error provides the PAE value for each residue pair (rounded to the nearest integer), and the field max_predicted_aligned_error gives the maximum possible PAE value, which is capped at 31.75 Å. The PAE is measured in Ångströms.
A separately developed 3D viewer of PAE allows for more intuitive visualization.[4]
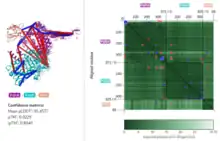
Interpretation
Interpretation of PAE values allows scientists to understand the level of confidence in the predicted structure of a protein: Lower PAE values between residue pairs from different domains indicate that the model predicts well-defined relative positions and orientations for those domains. Higher PAE values for such residue pairs suggest that the relative positions and/or orientations of these domains in the 3D structure are uncertain and should not be interpreted.[5]
Caveats
Although PAE provides valuable information, users should note that it is asymmetric; the PAE value for (x, y) may differ from the value for (y, x), particularly between loop regions with highly uncertain orientations.[6] Moreover, while AlphaFold can make useful inter-domain predictions, intra-domain prediction accuracy is expected to be more reliable based on CASP14 validation.
External links
References
- ↑ "AlphaFold Protein Structure Database". alphafold.ebi.ac.uk. 2023-06-12. Archived from the original on 2023-06-13. Retrieved 2023-06-12.
- ↑ "AlphaFold Error Estimates". www.rbvi.ucsf.edu. Archived from the original on 2023-06-13. Retrieved 2023-06-12.
- ↑ "Enabling high-accuracy protein structure prediction at the proteome scale". www.deepmind.com. 2023-06-13. Archived from the original on 2023-06-13. Retrieved 2023-06-13.
- ↑ Elfmann, Christoph; Stülke, Jörg (2023-05-04). "PAE viewer: a webserver for the interactive visualization of the predicted aligned error for multimer structure predictions and crosslinks". Nucleic Acids Research. 51 (W1): W404–W410. doi:10.1093/nar/gkad350. ISSN 0305-1048. PMC 10320053. PMID 37140053.
- ↑ Varadi, Mihaly (2023-06-13). "NIH: National Library of Medicine: AlphaFold Database". Nucleic Acids Research. 50 (D1): D439–D444. doi:10.1093/nar/gkab1061. PMC 8728224. PMID 34791371.
- ↑ "Why is the Alphafold PAE (predicted aligned error) not symmetric?". Matter Modeling Stack Exchange. 2023-06-12. Archived from the original on 2023-06-13. Retrieved 2023-06-12.