| Propionic acidemia | |
|---|---|
| Other names | Hyperglycinemia with ketoacidosis and leukopenia |
 | |
| Propionic acid | |
| Specialty | Endocrinology |
| Symptoms | Poor muscle tone, lethargy, vomiting |
| Diagnostic method | Genetic testing; high levels of propionic acid in the urine |
| Treatment | Low-protein diet |
| Prognosis | Development may be normal, or patients may have lifelong learning disabilities |
Propionic acidemia, also known as propionic aciduria or propionyl-CoA carboxylase deficiency (PCC deficiency),[1] is a rare autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia.[2][3]
The disorder presents in the early neonatal period with poor feeding, vomiting, lethargy, and lack of muscle tone.[4] Without treatment, death can occur quickly, due to secondary hyperammonemia, infection, cardiomyopathy, or brain damage.[5]
Symptoms and signs
Propionic acidemia can vary in severity.[6] Severe propionic acidemia lead to symptoms already seen in newborns.[7] Symptoms include poor feeding, vomiting, dehydration, acidosis, low muscle tone (hypotonia), seizures, and lethargy. The effects of propionic acidemia quickly become life-threatening.
Long-term complications can include chronic kidney disease,[8] cardiomyopathy, and prolonged QTc interval.[9]
Pathophysiology

In healthy individuals, enzyme propionyl-CoA carboxylase converts propionyl-CoA to methylmalonyl-CoA. This is one of many steps in the process of converting certain amino acids and fats into energy. Individuals with propionic acidemia cannot perform this conversion because the enzyme propionyl-CoA carboxylase is nonfunctional. The essential amino acids valine, methionine, isoleucine, and threonine can not be converted and this leads to a buildup of propionyl-CoA. Instead of being converted to methylmalonyl-CoA, propionyl-CoA is then converted into propionic acid, which builds up in the bloodstream. This in turn causes an accumulation of dangerous acids and toxins, which can cause damage to the organs.
In many cases, propionic acidemia can damage the brain, heart, kidney, liver, cause seizures and delays to normal development such as walking or talking. The accumulation of propionic acid is known to induce differential responses in different organs. The heart and liver are specific targets of the complication. The patient may need to be hospitalized to prevent breakdown of proteins within the body. Dietary needs must be closely managed.
Mutations in both copies of the PCCA or PCCB genes cause propionic acidemia.[10] These genes contain instructions to form alpha- and beta-subunits of PCC, the enzyme called propionyl-CoA carboxylase.
PCC is required for the normal breakdown of the essential amino acids valine, isoleucine, threonine, and methionine, as well as certain odd-chained fatty-acids. Mutations in the PCCA or PCCB genes disrupt the function of the enzyme, preventing these acids from being metabolized. As a result, propionyl-CoA, propionic acid, ketones, ammonia, and other toxic compounds accumulate in the blood, causing the signs and symptoms of propionic acidemia. Hyperammonemia develops due to the inhibitory effects of propionyl-CoA on N-acetylglutamate synthase, indirectly resulting in slowing of the urea cycle.[11]
Diagnosis
Elevated metabolites of propionic acid (for example, 3-hydroxypropionate, 2-methylcitrate, tiglylglycine, propionylglycine) found in blood and urine along with normal activity of biotinidase and normal levels of methylmalonic acid.[9]
Management
Patients with propionic acidemia should be started as early as possible on a low protein diet. In addition to a protein mixture that is devoid of methionine, threonine, valine, and isoleucine, the patient should also receive L-carnitine treatment and should be given antibiotics 10 days per month in order to remove the intestinal propiogenic flora. The patient should have diet protocols prepared for them with a “well day diet” with low protein content, a “half emergency diet” containing half of the protein requirements, and an “emergency diet” with no protein content. These patients are under the risk of severe hyperammonemia during infections that can lead to comatose states.[12]
Liver transplant is gaining a role in the management of these patients, with small series showing improved quality of life.
Epidemiology
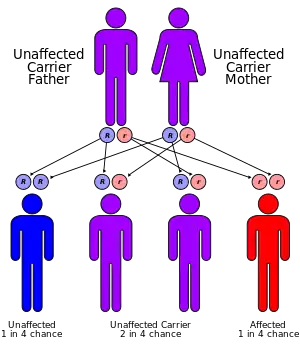
Propionic acidemia is inherited in an autosomal recessive pattern and is found in about 1 in 35,000[10] live births in the United States. The condition appears to be more common in Saudi Arabia,[13] with a frequency of about 1 in 3,000.[10] The condition also appears to be common in Amish, Mennonite and other populations with higher frequency of consanguinity.[14]
History
In 1957, a male child was born with poor mental development, repeated attacks of acidosis, and high levels of ketones and glycine in the blood. Upon dietary testing, Dr. Barton Childs discovered that his symptoms worsened when given the amino acids leucine, isoleucine, valine, methionine, and threonine. In 1961, the medical team at Johns Hopkins Hospital in Baltimore, Maryland published the case, calling the disorder ketotic hyperglycinemia. In 1969, using data from the original patient's sister, scientists established that propionic acidemia was a recessive disorder, and that propionic acidemia and methylmalonic acidemia are caused by deficiencies in the same enzyme pathway.[15]
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM): 606054
- ↑ Ravn K; Chloupkova M; Christensen E; Brandt NJ; Simonsen H; Kraus JP; Nielsen IM; Skovby F; Schwartz M (July 2000). "High incidence of propionic acidemia in greenland is due to a prevalent mutation, 1540insCCC, in the gene for the beta-subunit of propionyl CoA carboxylase". American Journal of Human Genetics. 67 (1): 203–206. doi:10.1086/302971. PMC 1287078. PMID 10820128.
- ↑ Deodato F, Boenzi S, Santorelli FM, Dionisi-Vici C (2006). "Methylmalonic and propionic aciduria". Am J Med Genet C Semin Med Genet. 142 (2): 104–112. doi:10.1002/ajmg.c.30090. PMID 16602092. S2CID 21114631.
- ↑ "Propionic acidemia". National Center for Advancing Translational Sciences. 2 Dec 2015. Retrieved 6 June 2018.
- ↑ Hamilton RL, Haas RH, Nyhan WC, Powell HC, Grafe MR (1995). "Neuropathology of propionic acidemia: a report of two patients with basal ganglia lesions". Journal of Child Neurology. 10 (1): 25–30. doi:10.1177/088307389501000107. PMID 7769173. S2CID 12674920.
- ↑ Shchelochkov, Oleg A.; Manoli, Irini; Juneau, Paul; Sloan, Jennifer L.; Ferry, Susan; Myles, Jennifer; Schoenfeld, Megan; Pass, Alexandra; McCoy, Samantha; Van Ryzin, Carol; Wenger, Olivia; Levin, Mark; Zein, Wadih; Huryn, Laryssa; Snow, Joseph (August 2021). "Severity modeling of propionic acidemia using clinical and laboratory biomarkers". Genetics in Medicine. 23 (8): 1534–1542. doi:10.1038/s41436-021-01173-2. ISSN 1530-0366. PMC 8354856. PMID 34007002. S2CID 234779025.
- ↑ Shchelochkov, Oleg A.; Carrillo, Nuria; Venditti, Charles (1993), Adam, Margaret P.; Everman, David B.; Mirzaa, Ghayda M.; Pagon, Roberta A. (eds.), "Propionic Acidemia", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 22593918, retrieved 2022-11-26
- ↑ Shchelochkov, Oleg A.; Manoli, Irini; Sloan, Jennifer L.; Ferry, Susan; Pass, Alexandra; Van Ryzin, Carol; Myles, Jennifer; Schoenfeld, Megan; McGuire, Peter; Rosing, Douglas R.; Levin, Mark D. (2019-06-28). "Chronic kidney disease in propionic acidemia". Genetics in Medicine. 21 (12): 2830–2835. doi:10.1038/s41436-019-0593-z. ISSN 1530-0366. PMC 7045176. PMID 31249402.
- 1 2 Shchelochkov, Oleg A.; Carrillo, Nuria; Venditti, Charles (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Propionic Acidemia", GeneReviews®, University of Washington, Seattle, PMID 22593918, retrieved 2019-09-29
- 1 2 3 http://mayoresearch.mayo.edu/mayo/research/barry_lab/ropionic-Aciademia.cfm Archived 2008-08-29 at the Wayback Machine
Barry Lab - Vector and Virus Engineering. Gene therapy for Propionic Acidemia - ↑ Dercksen, M.; IJlst, L.; Duran, M.; Mienie, L. J.; van Cruchten, A.; van der Westhuizen, F. H.; Wanders, R. J. A. (December 2014). "Inhibition of N-acetylglutamate synthase by various monocarboxylic and dicarboxylic short-chain coenzyme A esters and the production of alternative glutamate esters". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1842 (12 Pt A): 2510–2516. doi:10.1016/j.bbadis.2013.04.027. ISSN 0006-3002. PMID 23643712.
- ↑ Saudubray JM, Van Der Bergh G, Walter J : Inborn Metabolic Diseases Diagnosis and Treatment (2012)
- ↑ Al-Odaib AN, Abu-Amaro KK, Ozand PT, Al-Hellani AM (2003). "A new era for preventive genetic programs in the Arabian Peninsula". Saudi Medical Journal. 24 (11): 1168–1175. PMID 14647548.
- ↑ Kidd JR, Wolf B, Hsia E, Kidd KK (1980). "Genetics of propionic acidemia in a Mennonite-Amish kindred". Am J Hum Genet. 32 (2): 236–245. PMC 1686010. PMID 7386459.
- ↑ Hsia, T. (2003). "How Ketotic Hyperglycinemia Became Propionic Acidemia" (PDF). Paresearch.org. Retrieved 7 June 2018.
External links
- Propionic acidemia at NLM Genetics Home Reference
- Propionic acidemia at NIH's Office of Rare Diseases
- "Propionic acidemia". Orphanet.