In organic chemistry, α-halo ketones can be reduced with loss of the halogen atom to form enolates. The α-halo ketones are readily prepared from ketones by various ketone halogenation reactions, and the products are reactive intermediates that can be used for a variety of other chemical reactions.
Introduction
The reduction of α-halo ketones generates a variety of product structures that may exhibit unique substitution patterns and reactivity. For instance, reduction of α,α'-dihalo ketones leads to 2-oxyallyl metal complexes, which participate in [4+3] and [3+2] cycloaddition reactions as the 2π component.[1] 2-Oxyallyl metal intermediates may also intercept nucleophiles in a process that involves umpolung at the α carbon.[2] In addition, because reduction of monohalo ketones produces enolates in a site-specific fashion, reactions associated with enolates (alkylation, aldol, Michael) may be carried out using halo ketone substrates under reductive conditions.
(1)
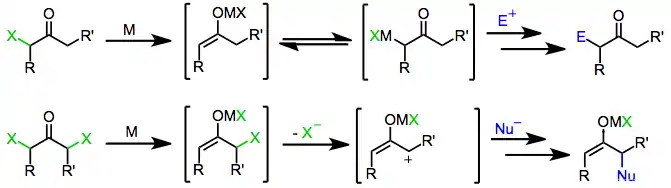
Mechanism
Many reducing agents used in this type of reaction are commercially available. A few require preparation and immediate use, including zinc–copper couple and zinc/silver couples.[3][4] and organocuprates[5]
Monohalo ketones
Monohalo ketones are reduced by both one-electron and two-electron reducing agents to afford the parent ketones, derivatives functionalized with electrophiles, or products of dimerization. The mechanism of reduction itself depends on the nature of the reducing agent.
One-electron reducing agents, such as d6 or d1 transition metal complexes, initially donate a single electron to the halo ketone. Fragmentation of the resulting radical anion yields an organic radical and halide anion. Donation of a second electron from a second equivalent of reducing agent leads to the formation of a metal enolate in which the oxidation state of the reducing agent has increased by one.[6]
(2)
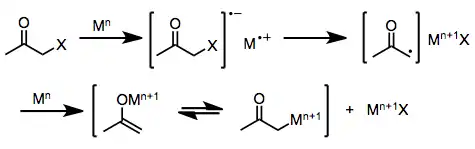
Two-electron reducing agents, the most prominent of which is zinc metal, undergo direct oxidative addition to monohalo ketones to afford metal enolates in which the oxidation state of the metal has increased by two. Subsequent disproportionation with unreacted halo ketone sometimes leads to the formation of two equivalents of enol radical, which may subsequently dimerize.[7]
(3)

Treatment of enolates derived from either one- or two-electron reducing agents with an electrophile affords α-functionalized ketones as the final product.
α,α-Dihalo ketones
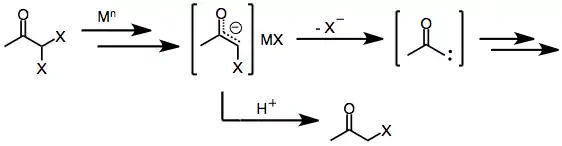
Similar to monohalo ketones, geminal dihalo ketones initially form metal enolates upon exposure to one- or two-electron reducing agents. In the presence of a protic solvent, reduction stops at this stage to afford the monohalo ketone (which may be further reduced to the parent ketone).[8] However, in the absence of a trapping electrophile or protic solvent, loss of the remaining halide from the enolate intermediate affords α-keto carbenes or carbenoids, which undergo C-H insertion reactions.[9]
(4)
α,α'-Dihalo ketones
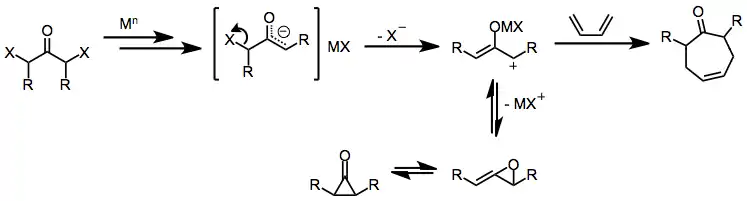
Initial reduction of α,α'-dihalo ketones leads to metal enolate intermediates. Loss of the remaining halide generates 2-oxyallyl metal complexes, which undergo [4+3] and [3+2] cycloaddition reactions in the presence of dienes or olefins.[1] Isomerization of 2-oxyallyl metal complexes to cyclopropanone and allene oxide forms is rapid and reversible; increasing the covalent character of the oxygen–metal bond favors the 2-oxyallyl isomer.[10]
(5)
Scope and Limitations
Reductions of α-halo ketones may afford the parent ketones, partially reduced halo ketones, or products arising from interception of intermediates along the reduction pathway with nucleophiles or electrophiles. The products obtained depend on both the reducing agent and the substitution pattern of the halo ketone.
Monohalo ketones
Monohalo ketones may be reduced to the corresponding parent ketone with lithium metal, followed by protonation of the enolate.[11] Other useful reducing agents for this transformation include lithium dialkylcuprates[12] and molybdenum hexacarbonyl-alumina.[13]
(6)
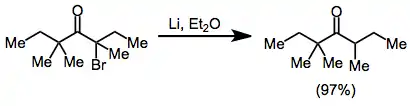
Alkylation of enolates generated through the reduction of monohalo ketones is limited to the most reactive alkyl halides.[14] However, reduction in the presence of an aldehyde leads to reductive aldol products, analogous to the Reformatsky reaction of haloesters. Zinc dust may be used alone; or as an amalgam, in combination with diethylaluminum chloride, or with catalytic amounts of copper(I) bromide.[15]
(7)
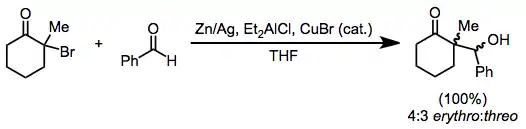
Reductive dimerization may result from the combination of two intermediate α-acyl radicals or nucleophilic attack of a metal enolate on unreacted halo ketone. Although complex reaction mixtures often result,[6] selective dimerization is possible in some cases. In the example below, the product distribution is solvent dependent[16]
(8)
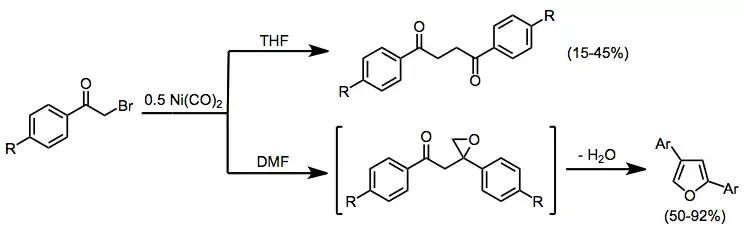
α,α-Dihalo ketones
Depending on the conditions employed, geminal dihalo ketones may be reduced to the parent ketone, monohalo ketone, or functionalized monohalo ketone with organocuprates.[17] Warming functionalized α-halo ketones produced under these conditions from −40 °C to room temperature leads to the corresponding α,β-unsaturated ketone.
(9)
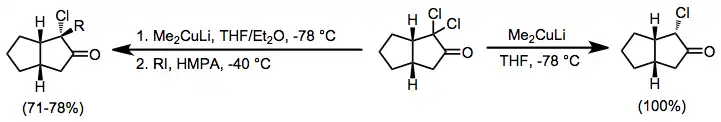
α,α'-Dihalo ketones
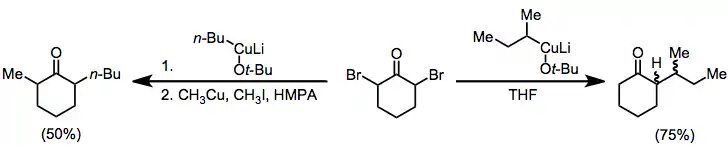
Organocuprates may also be employed for the reduction of α,α'-dihalo ketones to alkylated ketones. In the absence of an electrophile the monoalkyl ketone is isolated in good yield;[18] when an alkyl iodide is added, the dialkyl ketone is isolated (geminal alkylation is a problematic side reaction).[19]
(10)
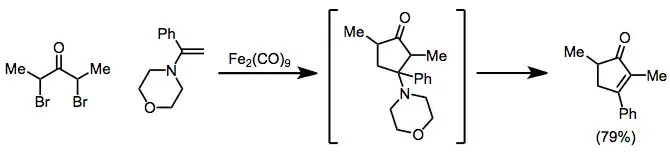
The 2-oxyallyl metal intermediates generated during reductions of α,α'-dihalo ketones with iron(0) complexes participate in [4+3] and [3+2] cycloaddition reactions to form cycloheptenones and cyclopentanones, respectively. During [3+2] cycloaddition reactions, the substituent on the alkene may eliminate to provide cyclopentenones from alkenes in one pot.[20]
(11)
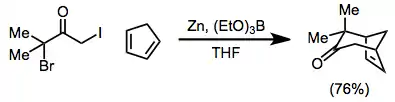
The scope of [4+3] cycloadditions is broad[1]—the reaction may be used to form cycloheptenones, as well as bridged[21] or fused[22] polycyclic products. Reduction may be carried out in the presence of the 4π component[23]
(12)
Synthetic applications
[4+3] cycloadditions of pyrroles may be used to establish the bicyclic skeleton of tropane alkaloids. For instance, a synthesis of scopine uses the [4+3] cycloaddition of N-methoxycarbonylpyrrole and α,α,α',α'-tetrabromoacetone as a key step. Diastereoselective reduction with diisobutylaluminum hydride (DIBAL-H) followed by epoxidation with trifluoroperacetic acid[24] affords the target.
(13)

References
- 1 2 3 Rigby, J.; Pigge, C. Org. React. 1997, 51, 351.
- ↑ Fry, J. ; O'Dea, J. J. Org. Chem. 1975, 40, 3625.
- ↑ LeGoff, E J. Org. Chem. 1964, 29, 2048.
- ↑ Denis, M. ; Girard, C. ; Conia, M. Synthesis, 1972, 549.
- ↑ Ho, L. ; Olah, A. Synthesis, 1976, 807.
- 1 2 Alper, H.; Keung, E. C. H J. Org. Chem. 1972, 37, 2566.
- ↑ Ghera, E. ; Perry, H. ; Shoua, S. J. Chem. Soc., Chem. Commun., 1973, 858.
- ↑ Greene, A. E.; Deprés, J.-P J. Am. Chem. Soc. 1979, 101, 4003.
- ↑ Scott, T. ; Cotton, D J. Am. Chem. Soc. 1973, 95, 2708.
- ↑ Bingham, C. ; Dewar, S. ; Lo, H J. Am. Chem. Soc. 1975, 97, 1302.
- ↑ Dubois, J.-E.; Fournier, P.; Lion, C. C. R. Acad. Sci., Ser. C 1974, 279, 965 (1974).
- ↑ Bull, J. R.; Tuinman, A. Tetrahedron Lett. 1973, 4349.
- ↑ Alper H.; Pattee, L. J. Org. Chem. 1979, 44, 2568.
- ↑ Dubois, E. ; Fournier, P. ; Lion, C C. R. Acad. Sci., Ser. C 1974, 279, 965.
- ↑ Maruoka, K. ; Hashimoto, S. ; Kitagawa, Y. ; Yamamoto, H. ; Nozaki, H J. Am. Chem. Soc. 1977, 99, 7705.
- ↑ Yoshisato, E. ; Tsutsumi, S J. Am. Chem. Soc. 1968, 90, 4488.
- ↑ Deprés, J.-P.; Greene, A. E. J. Org. Chem. 1980, 45, 2036.
- ↑ Posner, H. ; Sterling, J J. Am. Chem. Soc. 1973, 95, 3076.
- ↑ Posner, H. ; Sterling, J. ; Whitten, E. ; Lentz, M. ; Brunelle, J J. Am. Chem. Soc. 1975, 97, 107.
- ↑ Hayakawa, Y.; Yokoyama, K.; Noyori, R. J. Am. Chem. Soc. 1978, 100, 1799.
- ↑ Noyori, R.; Nishizawa, M.; Shimizu, F.; Hayakawa, Y.; Maruoka, K.; Hashimoto, S.; Yamamoto, H.; Nozaki, H J. Am. Chem. Soc. 1979, 101, 220.
- ↑ Hayakawa, Y. ; Yokoyama, K. ; Noyori, R J. Am. Chem. Soc. 1978, 100, 1799.
- ↑ Hoffmann, R. ; Iqbal, N. Tetrahedron Lett., 1975, 4487.
- ↑ Hayakawa, Y.; Baba, Y.; Makino, S.; Noyori, R. J. Am. Chem. Soc. 1978, 100, 1786.