STARR-seq (short for self-transcribing active regulatory region sequencing) is a method to assay enhancer activity for millions of candidates from arbitrary sources of DNA. It is used to identify the sequences that act as transcriptional enhancers in a direct, quantitative, and genome-wide manner.[1]
In eukaryotes, transcription is regulated by sequence-specific DNA-binding proteins (transcription factors) associated with a gene’s promoter and also by distant control sequences including enhancers. Enhancers are non-coding DNA sequences, containing several binding sites for a variety of transcription factors.[2] They typically recruit transcriptional factors that modulate chromatin structure and directly interact with the transcription machinery placed at the promoter of a gene. Enhancers are able to regulate transcription of target genes in a cell type-specific manner,[1] independent of their location or distance from the promoter of genes. In certain contexts (see Transvection (genetics)), they can even regulate transcription of genes located in a different chromosome.[3] However, the knowledge about enhancers so far has been limited to studies of a small number of enhancers, as they have been difficult to identify accurately at a genome-wide scale.[2] Moreover, many regulatory elements function only in certain cell types and specific conditions.[4]
Enhancer detection
Enhancer detection in Drosophila is an original methodology using random insertion of transposon-derived vector that encodes a reporter protein downstream of a minimal promoter. This approach allows to observe the expression of reporter in transgenic animals and provides information about nearby genes that are regulated by these sequences. The discovery and characterization of cell types along with genes involved in their determination have been significantly improved by the discovery of this technique.[5][6][7][8]
During the past few years, post-genomic technologies, have displayed specific features of poised and active enhancers that have improved enhancer discovery.[2] Development of new methods such as deep sequencing of DNase I hypersensitive sites (DNase-Seq), formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-Seq), chromatin immunoprecipitation followed by deep sequencing (ChIP-sequencing), and MNase-defined cistrome-Occupancy Analysis (MOA-seq[9]), provide genome-wide enhancer predictions by enhancer-associated chromatin features.[1]
Application
However, DNase-seq and FAIRE-seq alone fail to provide a direct functional or quantitative readout of enhancer activity, so reporter assays that can deduce enhancer strength from the quantitative enrichment of reporter transcripts are needed to assess enhancer activity quantitatively. Yet, these assays are not high-throughput (High throughput biology), as it is impossible to conduct millions of tests required for identification of enhancers in a genome-wide manner.[1] The development of STARR-seq attempts to circumvent this analytical barrier. Taking advantage of the knowledge that enhancers can work independently of their relative locations, candidate sequences are placed downstream of a minimal promoter, allowing the active enhancers to transcribe themselves. The strength of each enhancer is then reflected by its relative enrichment among cellular RNAs. Such a direct coupling of candidate sequences to enhancer activity enables the parallel evaluation of millions of DNA fragments from arbitrary sources.[1]
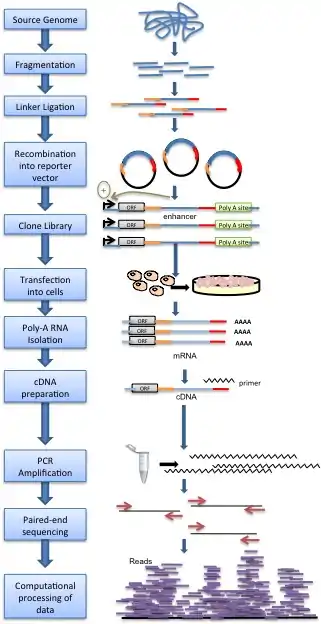
Methodology
Genomic DNA is randomly sheared and broken down to small fragments. Adaptors are ligated to size-selected DNA fragments. Next, adaptor-linked fragments are amplified and the PCR products are purified followed by placing candidate sequences downstream of a minimal promoter of screening vectors, giving them an opportunity to transcribe themselves. Candidate cells are then transfected with reporter library and cultured. Thereafter, total RNAs are extracted and poly-A RNAs isolated. Using reverse transcription method, cDNAs are produced, amplified and then candidate fragments are used for high-throughput paired end sequencing. Sequence reads are mapped to the reference genome and computational processing of data is carried out.[1]
Identification of enhancers
Applying this technology to Drosophila genome, Arnold et al.[1] found 96% of the non-repetitive genome with at least 10-fold coverage. Authors discovered that most identified enhancers (55.6%) were placed within introns, particularly in the first intron and intergenic regions. 4.5% of enhancers were located at transcription start sites (TSS), suggesting that these enhancers can start transcription and also improve transcription from a distant TSS.[1] The strongest enhancers were near housekeeping genes such as enzymes or component of the cytoskeleton and developmental regulators such as the transcription factors. The strongest enhancer was located within the intron of the transcription factor zfh1. This transcription factor regulates neuropeptide expression and growth of larval neuromuscular junctions in Drosophila.[10] The ribosomal protein genes were the only class of genes with poor enhancers ranking. Moreover, authors demonstrated that many genes are regulated by several independent active enhancers even in a single cell type. Furthermore, gene expression levels on average were correlated with the sum of the enhancer strengths per gene, supporting direct link between gene expression and enhancer activity.[1]
Characterization of variant alleles
Applying this technology to the characterization and discovery of regulatory variant alleles, Vockley et al.[11] characterized the effects of human genetic variation on non-coding regulatory element function, measuring the activity of 100 putative enhancers captured directly from the genomes of 95 members of a study cohort. This approach enables the functional fine-mapping of causal regulatory variants in regions of high linkage disequilibrium identified by eQTL analyses. This approach provides a general path forward to identify perturbations in gene regulatory elements that contribute to complex phenotypes.
Quantifying enhancer activity
STARR-seq has been used to measure the regulatory activity of DNA fragments that have been enriched for sites occupied by specific transcription factors. Cloning ChIP DNA libraries generated from chromatin immunoprecipitation of the glucocorticoid receptor into STARR-seq enabled genome-scale quantification of glucocorticoid-induced enhancer activity.[12] This approach is useful for measuring the differences in enhancer activity between sites that are bound by the same transcription factor.
Advantages
- A quantitative genome-wide assay for enhancer detection.[1]
- Applicable technique for screening arbitrary sources of DNA in any cell type or tissue that allows adequate introduction of reporter constructs.[1]
- A method with high detection rate (>99%) by employing pair-end sequencing, even for sequences that contain transcript-destabilizing elements.
- Technique to evaluate the strength of enhancers quantitatively, and identify endogenously silenced enhancers by integrating them into a chromosomal context.[1]
Future directions
By combining traditional approach with high-throughput sequencing technology and highly specialized bio-computing methods, STARR-seq is able to detect enhancers in a quantitative and genome-wide manner. The study of gene regulation and their responsible pathways in the genome during normal development and also in disease can be very demanding. Therefore, applying STARR-seq to many cell types across organisms supports identifying cell type-specific gene regulatory elements and practically assesses non-coding mutations causing disease. Recently, a related approach coupling capture of regions of interest to STARR-seq technique have been developed and extensively validated in mammalian cell lines.[13]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A (March 2013). "Genome-wide quantitative enhancer activity maps identified by STARR-seq". Science. 339 (6123): 1074–1077. Bibcode:2013Sci...339.1074A. doi:10.1126/science.1232542. PMID 23328393. S2CID 54488955.
- 1 2 3 Xu J, Smale ST (November 2012). "Designing an enhancer landscape". Cell. 151 (5): 929–931. doi:10.1016/j.cell.2012.11.007. PMC 3732118. PMID 23178114.
- ↑ Ong CT, Corces VG (April 2011). "Enhancer function: new insights into the regulation of tissue-specific gene expression". Nature Reviews. Genetics. 12 (4): 283–293. doi:10.1038/nrg2957. PMC 3175006. PMID 21358745.
- ↑ Baker M (May 2011). "Highlighting enhancers". Nature Methods. 8 (5): 373. doi:10.1038/nmeth0511-373. PMID 21678620.
- ↑ Bellen HJ (December 1999). "Ten years of enhancer detection: lessons from the fly". The Plant Cell. 11 (12): 2271–2281. doi:10.2307/3870954. JSTOR 3870954. PMC 144146. PMID 10590157.
- ↑ Bier E, Vaessin H, Shepherd S, Lee K, McCall K, Barbel S, et al. (September 1989). "Searching for pattern and mutation in the Drosophila genome with a P-lacZ vector". Genes & Development. 3 (9): 1273–1287. doi:10.1101/gad.3.9.1273. PMID 2558049.
- ↑ Wilson C, Pearson RK, Bellen HJ, O'Kane CJ, Grossniklaus U, Gehring WJ (September 1989). "P-element-mediated enhancer detection: an efficient method for isolating and characterizing developmentally regulated genes in Drosophila". Genes & Development. 3 (9): 1301–1313. doi:10.1101/gad.3.9.1301. PMID 2558051.
- ↑ O'Kane CJ, Gehring WJ (December 1987). "Detection in situ of genomic regulatory elements in Drosophila". Proceedings of the National Academy of Sciences of the United States of America. 84 (24): 9123–9127. Bibcode:1987PNAS...84.9123O. doi:10.1073/pnas.84.24.9123. PMC 299704. PMID 2827169.
- ↑ Savadel SD, Hartwig T, Turpin ZM, Vera DL, Lung PY, Sui X, et al. (August 2021). "The native cistrome and sequence motif families of the maize ear". PLOS Genetics. 17 (8): e1009689. doi:10.1371/journal.pgen.1009689. PMC 8360572. PMID 34383745.
- ↑ Vogler G, Urban J (July 2008). "The transcription factor Zfh1 is involved in the regulation of neuropeptide expression and growth of larval neuromuscular junctions in Drosophila melanogaster". Developmental Biology. 319 (1): 78–85. doi:10.1016/j.ydbio.2008.04.008. PMID 18499094.
- ↑ Vockley CM, Guo C, Majoros WH, Nodzenski M, Scholtens DM, Hayes MG, et al. (August 2015). "Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort". Genome Research. 25 (8): 1206–1214. doi:10.1101/gr.190090.115. PMC 4510004. PMID 26084464.
- ↑ Vockley CM, D'Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, et al. (August 2016). "Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome". Cell. 166 (5): 1269–1281.e19. doi:10.1016/j.cell.2016.07.049. PMC 5046229. PMID 27565349.
- ↑ Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LT, Fernandez N, et al. (April 2015). "High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq". Nature Communications. 6: 6905. Bibcode:2015NatCo...6.6905V. doi:10.1038/ncomms7905. PMID 25872643. S2CID 205336994.