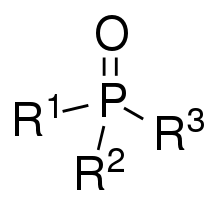
Phosphine oxides are phosphorus compounds with the formula OPX3. When X = alkyl or aryl, these are organophosphine oxides. Triphenylphosphine oxide is an example. An inorganic phosphine oxide is phosphoryl chloride (POCl3).[1]
Parent compound
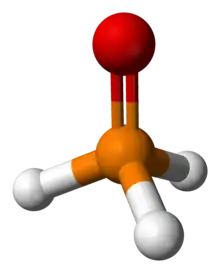
The parent compound phosphine oxide (H3PO) is unstable. It has been detected with mass spectrometry as a reaction product of oxygen and phosphine,[2] by means of FT-IR in a phosphine-ozone reaction[3] and in matrix isolation with a reaction of phosphine, vanadium oxytrichloride and chromyl chloride.[4] It has also been reported relatively stable in a water-ethanol solution by electrochemical oxidation of white phosphorus, where it slowly disproportionates into phosphine and hypophosphorous acid.[5] Secondary phosphine oxides (R2P(O)H) are tautomers of phosphinous acids (R2POH).
Phosphine oxide is reported as an intermediate in the room-temperature polymerization of phosphine and nitric oxide to solid PxHy.[6]
Structure and bonding
Tertiary phosphine oxides
Tertiary phosphine oxides are the most commonly encountered phosphine oxides. With the formula R3PO, they are tetrahedral compounds. They are usually prepared by oxidation of tertiary phosphines. The P-O bond is short and polar. According to molecular orbital theory, the short P–O bond is attributed to the donation of the lone pair electrons from oxygen p-orbitals to the antibonding phosphorus-carbon bonds.[7] The nature of the P–O bond was once hotly debated. Some discussions invoked a role for phosphorus-centered d-orbitals in bonding, but this analysis is not supported by computational analyses. In terms of simple Lewis structure, the bond is more accurately represented as a dative bond, as is currently used to depict an amine oxide.[8][9]
Secondary phosphine oxides
Secondary phosphine oxides (SPOs), formally derived from secondary phosphines (R2PH), are again tetrahedral at phosphorus.[10] One commercially available example of a secondary phosphine oxide is diphenylphosphine oxide. SPOs are used in the formulation of catalysts for cross coupling reactions.[11]
Unlike tertiary phosphine oxides, SPOs often undergo further oxidation, which enriches their chemistry:
- R2P(O)H + H2O2 → R2P(O)OH + H2O
These reactions are preceded by tautomerization to the phosphinous acid (R2POH):
- R2P(O)H R2POH

Primary phosphine oxides
Primary phosphine oxides, formally oxidized derivatives of primary phosphines, are again tetrahedral at phosphorus. With four different substituents (O, OH, H, R) they are chiral. The primary phosphine oxides subject to tautomerization, which leads to racemization, and further oxidation, analogous to the behavior of SPOs. Additionally, primary phosphine oxides are susceptible to disproportionation to the phosphinic acid and the primary phosphine:[12]
- 2 RP(O)H2 → RP(O)(H)OH + 2 RPH2
Syntheses
Phosphine oxide are typically produced by oxidation of organophosphines. The oxygen in air is often sufficiently oxidizing to fully convert trialkylphosphines to their oxides at room temperature:
- R3P + 1/2 O2 → R3PO
This conversion is usually undesirable. In order to suppress this reaction, air-free techniques are often employed when handling say, trimethylphosphine.
Less basic phosphines, such as methyldiphenylphosphine are converted to their oxides by treatment with hydrogen peroxide:[13]
- PMePh2 + H2O2 → OPMePh2 + H2O
Phosphine oxides are generated as a by-product of the Wittig reaction:
- R3PCR'2 + R"2CO → R3PO + R'2C=CR"2
Another albeit unconventional route to phosphine oxides is the thermolysis of phosphonium hydroxides:
- [PPh4]Cl + NaOH → Ph3PO + NaCl + PhH
The hydrolysis of phosphorus(V) dihalides also affords the oxide:[14]
- R3PCl2 + H2O → R3PO + 2 HCl
A special nonoxidative route is applicable secondary phosphine oxides, which arise by the hydrolysis of the chlorophosphine. An example is the hydrolysis of chlorodiphenylphosphine to give diphenylphosphine oxide:
- Ph2PCl + H2O → Ph2P(O)H + HCl
Deoxygenation
The deoxygenation of phosphine oxides has been extensively developed because many useful stoichiometric reactions convert tertiary phosphines to the corresponding oxides. Regeneration of the tertiary phosphine requires cheap oxophilic reagents, which are usually silicon-based. These deoxygenation reactions can be subdivided into stoichiometric and catalytic processes.[15]
Stoichiometric processes
Use of trichlorosilane is a standard laboratory method. Industrial routes use phosgene or equivalent reagents, which produce chlorotriphenylphosphonium chloride, which is separately reduced.[16] For chiral phosphine oxides, deoxygenation can proceed with retention or inversion of configuration. Classically, inversion is favored by a combination of trichlorosilane and triethylamine, whereas in the absence of the Lewis base, the reaction proceeds with retention.[17]
- HSiCl3 + Et3N ⇋ SiCl3− + Et3NH+
- R3PO + Et3NH+ ⇋ R3POH+ + Et3N
- SiCl3− + R3POH+ → PR3 + HOSiCl3
The popularity of this method is partly attributable to the availability of inexpensive trichlorosilane. Instead of HSiCl3, other perchloropolysilanes, e.g. hexachlorodisilane (Si2Cl6), can also be used. In comparison, using the reaction of the corresponding phosphine oxides with perchloropolysilanes such as Si2Cl6 or Si3Cl8 in benzene or chloroform, phosphines can be prepared in higher yields.
- R3PO + Si2Cl6 → R3P + Si2OCl6
- 2 R3PO + Si3Cl8 → 2 R3P + Si3O2Cl8
Deoxygenation has been effected with boranes and alanes.[15]
Catalytic processes
Phosphoric acids ((RO)2PO2H) catalyze the deoxygenation of phosphine oxides by hydrosilanes.[18]
Use
Phosphine oxides are ligands in various applications of homogeneous catalysis. In coordination chemistry, they are known to have labilizing effects to CO ligands cis to it in organometallic reactions. The cis effect describes this process.
References
- ↑ D. E. C. Corbridge "Phosphorus: An Outline of its Chemistry, Biochemistry, and Technology" 5th Edition Elsevier: Amsterdam 1995. ISBN 0-444-89307-5.
- ↑ Hamilton, Peter A.; Murrells, Timothy P. (1985). "Kinetics and mechanism of the reactions of PH3 with O(3P) and N(4S) atoms". J. Chem. Soc., Faraday Trans. 2 (81): 1531–1541. doi:10.1039/F29858101531.
- ↑ Withnall, Robert; Andrews, Lester (1987). "FTIR spectra of the photolysis products of the phosphine-ozone complex in solid argon". J. Phys. Chem. 91 (4): 784–797. doi:10.1021/j100288a008.
- ↑ Kayser, David A.; Ault, Bruce S. (2003). "Matrix Isolation and Theoretical Study of the Photochemical Reaction of PH3 with OVCl3 and CrCl2O2". J. Phys. Chem. A. 107 (33): 6500–6505. Bibcode:2003JPCA..107.6500K. doi:10.1021/jp022692e.
- ↑ Yakhvarov, D.; Caporali, M.; Gonsalvi, L.; Latypov, S.; Mirabello, V.; Rizvanov, I.; Sinyashin, O.; Stoppioni, P.; Peruzzini, M. (2011). "Experimental Evidence of Phosphine Oxide Generation in Solution and Trapping by Ruthenium Complexes". Angewandte Chemie International Edition. 50 (23): 5370–5373. doi:10.1002/anie.201100822. PMID 21538749.
- ↑ Zhao, Yi-Lei; Flora, Jason W.; David Thweatt, William; Garrison, Stephen L.; Gonzalez, Carlos; Houk, K. N.; Marquez, Manuel (2009). "Phosphine Polymerization by Nitric Oxide: Experimental Characterization and Theoretical Predictions of Mechanism". Inorg. Chem. 48 (3): 1223–1231. doi:10.1021/ic801917a. PMID 19102679.
- ↑ D. B. Chesnut (1999). "The Electron Localization Function (ELF) Description of the PO Bond in Phosphine Oxide". Journal of the American Chemical Society. 121 (10): 2335–2336. doi:10.1021/ja984314m.
- ↑ Gilheany, Declan G. (1994). "No d Orbitals but Walsh Diagrams and Maybe Banana Bonds: Chemical Bonding in Phosphines, Phosphine Oxides, and Phosphonium Ylides". Chemical Reviews. 94 (5): 1339–1374. doi:10.1021/cr00029a008. PMID 27704785.
- ↑ In fact, the N-O bonds in amine oxides are more likely to be closer to double bonds than are those of the P-O bonds in phosphine oxides; see e.g. https://pubs.rsc.org/en/content/articlelanding/2015/sc/c5sc02076j#:~:text=Quantitative%20analysis%20of%20known%20species%20of%20general%20formulae,high%20degree%20of%20covalent%20rather%20than%20ionic%20bonding.
- ↑ Gallen, Albert; Riera, Antoni; Verdaguer, Xavier; Grabulosa, Arnald (2019). "Coordination Chemistry and Catalysis with Secondary Phosphine oxides". Catalysis Science & Technology. 9 (20): 5504–5561. doi:10.1039/C9CY01501A. hdl:2445/164459. S2CID 202885438.
- ↑ Ackermann, Lutz (2007). "Catalytic Arylations with Challenging Substrates: From Air-Stable HASPO Preligands to Indole Syntheses and C-H-Bond Functionalizations". Synlett. 2007 (4): 0507–0526. doi:10.1055/s-2007-970744.
- ↑ Horký, Filip; Císařová, Ivana; Štěpnička, Petr (2021). "A Stable Primary Phosphane Oxide and Its Heavier Congeners". Chemistry – A European Journal. 27 (4): 1282–1285. doi:10.1002/chem.202003702. PMID 32846012. S2CID 221346479.
- ↑ Denniston, Michael L.; Martin, Donald R. (1977). Methyldiphenylphosphine Oxide and Dimethylphenylphosphine Oxide. Inorganic Syntheses. Vol. 17. pp. 183–185. doi:10.1002/9780470132487.ch50. ISBN 9780470132487.
- ↑ W. B. McCormack (1973). "3-Methyl-1-Phenylphospholene oxide". Organic Syntheses.; Collective Volume, vol. 5, p. 787
- 1 2 Podyacheva, Evgeniya; Kuchuk, Ekaterina; Chusov, Denis (2019). "Reduction of phosphine oxides to phosphines". Tetrahedron Letters. 60 (8): 575–582. doi:10.1016/j.tetlet.2018.12.070. S2CID 104364715.
- ↑ van Kalkeren, Henri A.; van Delft, Floris L.; Rutjes, Floris P. J. T. (2013). "Organophosphorus Catalysis to Bypass Phosphine Oxide Waste". ChemSusChem. 6 (9): 1615–1624. doi:10.1002/cssc.201300368. hdl:2066/117145. ISSN 1864-5631. PMID 24039197.
- ↑ Klaus Naumann; Gerald Zon; Kurt Mislow (1969). "Use of hexachlorodisilane as a reducing agent. Stereospecific deoxygenation of acyclic phosphine oxides". Journal of the American Chemical Society. 91 (25): 7012–7023. doi:10.1021/ja01053a021.
- ↑ Li, Yuehui; Lu, Liang-Qiu; Das, Shoubhik; Pisiewicz, Sabine; Junge, Kathrin; Beller, Matthias (2012). "Highly Chemoselective Metal-Free Reduction of Phosphine Oxides to Phosphines". Journal of the American Chemical Society. 134 (44): 18325–18329. doi:10.1021/ja3069165. ISSN 0002-7863. PMID 23062083.