| Spinal muscular atrophies | |
|---|---|
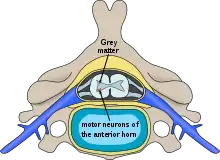 | |
| Location of neurons affected in spinal muscular atrophies | |
| Specialty | Neurology |
| Symptoms | Loss of motor neurons resulting in muscle wasting |
Spinal muscular atrophies (SMAs) are a genetically and clinically heterogeneous group of rare debilitating disorders characterised by the degeneration of lower motor neurons (neuronal cells situated in the anterior horn of the spinal cord) and subsequent atrophy (wasting) of various muscle groups in the body.[1] While some SMAs lead to early infant death, other diseases of this group permit normal adult life with only mild weakness.
Classification
Based on the type of muscles affected, spinal muscular atrophies can be divided into:
- Proximal spinal muscular atrophies, i.e., conditions that affect primarily proximal muscles;
- Distal spinal muscular atrophies (which significantly overlap with distal hereditary motor neuronopathies) where they affect primarily distal muscles.
When taking into account prevalence, spinal muscular atrophies are traditionally divided into:
- Autosomal recessive proximal spinal muscular atrophy, responsible for 90-95% of cases and usually called simply spinal muscular atrophy (SMA) – a disorder associated with a genetic mutation on the SMN1 gene on chromosome 5q (locus 5q13), diagnosed predominantly in young children and in its most severe form being the most common genetic cause of infant death if left untreated;
- Localised spinal muscular atrophies – much more rare conditions, in some instances described in but a few patients in the world, which are associated with mutations of genes other than SMN1 and for this reason sometimes termed simply non-5q spinal muscular atrophies; none has currently a causal treatment.
A more detailed classification is based on the gene associated with the condition (where identified) and is presented in table below.
| Group | Name Alternative names |
OMIM | Gene | Locus | Mode of inheritance |
Characteristics |
|---|---|---|---|---|---|---|
| SMA | Spinal muscular atrophy (SMA)
|
253300 253550 253400 271150 |
SMN1 | 5q13.2 | Autosomal recessive | Affects primarily proximal muscles in people of all ages, progressive, relatively common |
| XLSMA | X-linked spinal muscular atrophy type 1 (SMAX1)
|
313200 | NR3C4 | Xq12 | X-linked recessive | Affects primarily bulbar muscles as well as sensory nerves mainly in adult men, progressive |
X-linked spinal muscular atrophy type 2 (SMAX2)
|
301830 | UBA1 | Xp11.23 | X-linked recessive | Characterised by bone fractures, affects mainly distal muscles in newborn boys, usually fatal in infancy | |
X-linked spinal muscular atrophy type 3 (SMAX3)
|
300489 | ATP7A | Xq21.1 | X-linked recessive | Affects distal muscles of all extremities mainly in boys, slowly progressive | |
| DSMA | Distal spinal muscular atrophy type 1 (DSMA1)
|
604320 | IGHMBP2 | 11q13.3 | Autosomal recessive | Affects mainly infant boys, similar to SMA type 1 but with diaphragmatic paralysis |
Distal spinal muscular atrophy type 2 (DSMA2)
|
605726 | SIGMAR1 | 19p13.3 | Autosomal recessive | Slowly progressive | |
Distal spinal muscular atrophy type 3 (DSMA3)
|
607088 | ? | 11q13.3 | Autosomal recessive | Slowly progressive | |
| Distal spinal muscular atrophy type 4 (DSMA4) | 611067 | PLEKHG5 | 1p36.31 | Autosomal recessive | Slowly progressive, described only in one family | |
| Distal spinal muscular atrophy type 5 (DSMA5) | 614881 | DNAJB2 | 2q35 | Autosomal recessive | Young adult onset, slowly progressive | |
Distal spinal muscular atrophy type VA (DSMAVA)
|
600794 | GARS | 7p14.3 | Autosomal dominant | With upper limb predominance; allelic and overlapping with CMT2D, phenotype overlapping with Silver syndrome | |
Distal spinal muscular atrophy type VB (DSMAVB)
|
614751 | REEP1 | 2p11 | Autosomal dominant | With upper limb predominance; allelic and overlapping with HSP-31 | |
Distal spinal muscular atrophy with calf predominance
|
615575 | FBXO38 | 5q32 | Autosomal dominant | Juvenile- or adult-onset, slowly progressive, affects both proximal and distal muscles, initially manifests with calf weakness which progresses to hands | |
Distal spinal muscular atrophy with vocal cord paralysis
|
158580 | SLC5A7 | 2q12.3 | Autosomal dominant | Adult-onset with vocal cord paralysis, very rare | |
Congenital distal spinal muscular atrophy
|
600175 | TRPV4 | 12q24.11 | Autosomal dominant | Affects primarily distal muscles of lower limbs, non-progressive, rare, allelic with SPSMA and CMT2C | |
Scapuloperoneal spinal muscular atrophy (SPSMA)
|
181405 | TRPV4 | 12q24.11 | Autosomal dominant or X-linked dominant |
Affects muscles of lower limbs, non-progressive, rare, allelic with congenital distal spinal muscular atrophy and CMT2C | |
Autosomal dominant distal spinal muscular atrophy
|
158590 | HSPB8 | 12q24.23 | Autosomal dominant | Adult-onset. Allelic with Charcot–Marie–Tooth disease type 2L (CMT2L) | |
Autosomal dominant juvenile distal spinal muscular atrophy
|
182960 | ? | 7q34–q36 | Autosomal dominant | Juvenile-onset | |
| Juvenile segmental spinal muscular atrophy (JSSMA) | 183020 | ? | 18q21.3 | ? | Juvenile-onset, progressive with stabilisation after 2–4 years, affects primarily hands, very rare | |
| Finkel type proximal spinal muscular atrophy (SMAFK) | 182980 | VAPB | 20q13.32 | Autosomal dominant | Late-onset, affects proximal muscles in adults | |
| James type infantile spinal muscular atrophy (SMAJI) | 619042 | GARS1 | 7p14.3 | Autosomal dominant | Infantile-onset hypotonia, slowly progressive, resulting in delayed motor milestones and loss of previous motor skills. Children never walk. Milder disorders caused by GARS1 mutations are CMT2D and HMN5A. | |
| Jokela type spinal muscular atrophy (SMAJ) | 615048 | CHCHD10 | 22q11.2–q13.2 | Autosomal dominant | Late-onset, slowly progressive, affects both proximal and distal muscles in adults | |
| Spinal muscular atrophy with lower extremity predominance 1 (SMALED1) | 158600 | DYNC1H1 | 14q32 | Autosomal dominant | Affects proximal muscles in infants | |
| Spinal muscular atrophy with lower extremity predominance 2A (SMALED2A) | 615290 | BICD2 | 9q22.31 | Autosomal dominant | Early-onset, primarily affecting lower limbs, slowly progressive, non-life-limiting, very rare | |
| Spinal muscular atrophy with lower extremity predominance 2B (SMALED2B) | 618291 | BICD2 | 9q22.31 | Autosomal dominant | Presents with hypotonia, contractures and respiratory involvement at birth, frequently fatal in early childhood, very rare | |
| Spinal muscular atrophy with progressive myoclonic epilepsy (SMAPME) | 159950 | ASAH1 | 8p22 | Autosomal recessive | ||
| Spinal muscular atrophy with congenital bone fractures 1 (SMABF1) | 616866 | TRIP4 | 15q22.31 | Autosomal recessive | Prenatal onset, characterised by severe muscle wasting, respiratory and feeding failure, and bone fractures at birth as in arthrogryposis multiplex congenita, usually fatal in infancy | |
| Spinal muscular atrophy with congenital bone fractures 2 (SMABF2) | 616867 | ASCC1 | 10q22.1 | Autosomal recessive | Prenatal onset, characterised by severe muscle wasting, respiratory and feeding failure, and bone fractures at birth as in arthrogryposis multiplex congenita, usually fatal in infancy[2][3][4] | |
| PCH | Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH)
|
607596 | VRK1 | 14q32 | Autosomal dominant | → see Pontocerebellar hypoplasia |
| MMA | Juvenile asymmetric segmental spinal muscular atrophy (JASSMA)
|
602440 | ? | ? | ? | → see Monomelic amyotrophy |
| PMA | Progressive spinal muscular atrophy
|
? | ? | ? | ? | → see Progressive muscular atrophy |
In all forms of SMA (with an exception of X-linked spinal muscular atrophy type 1), only motor neurons, located at the anterior horn of spinal cord, are affected; sensory neurons, which are located at the posterior horn of spinal cord, are not affected. By contrast, hereditary disorders that cause both weakness due to motor denervation along with sensory impairment due to sensory denervation are known as hereditary motor and sensory neuropathies (HMSN).
See also
References
- ↑ "Spinal muscular atrophy". Genetics Home Reference. 2016-03-21. Retrieved 2016-03-26.
- ↑ Knierim E, Hirata H, Wolf NI, Morales-Gonzalez S, Schottmann G, Tanaka Y, et al. (March 2016). "Mutations in Subunits of the Activating Signal Cointegrator 1 Complex Are Associated with Prenatal Spinal Muscular Atrophy and Congenital Bone Fractures". American Journal of Human Genetics. 98 (3): 473–489. doi:10.1016/j.ajhg.2016.01.006. PMC 4800037. PMID 26924529.
- ↑ Oliveira J, Martins M, Pinto Leite R, Sousa M, Santos R (October 2017). "The new neuromuscular disease related with defects in the ASC-1 complex: report of a second case confirms ASCC1 involvement". Clinical Genetics. 92 (4): 434–439. doi:10.1111/cge.12997. PMID 28218388. S2CID 28768062.
- ↑ Giuffrida MG, Mastromoro G, Guida V, Truglio M, Fabbretti M, Torres B, et al. (December 2019). "A new case of SMABF2 diagnosed in stillbirth expands the prenatal presentation and mutational spectrum of ASCC1". American Journal of Medical Genetics. Part A. 182 (3): 508–512. doi:10.1002/ajmg.a.61431. PMID 31880396. S2CID 209490732.
Further reading
- Van Den Berg-Vos RM, Van Den Berg LH, Visser J, de Visser M, Franssen H, Wokke JH (November 2003). "The spectrum of lower motor neuron syndromes". Journal of Neurology. 250 (11): 1279–92. doi:10.1007/s00415-003-0235-9. PMID 14648143. S2CID 25844355.
- Guillot N, Cuisset JM, Cuvellier JC, Hurtevent JF, Joriot S, Vallee L (March 2008). "Unusual clinical features in infantile Spinal Muscular Atrophies". Brain & Development. 30 (3): 169–78. doi:10.1016/j.braindev.2007.07.008. PMID 17804187. S2CID 24657851.