In organic chemistry, a sulfoxide, also called a sulphoxide, is an organosulfur compound containing a sulfinyl (>SO) functional group attached to two carbon atoms. It is a polar functional group. Sulfoxides are oxidized derivatives of sulfides. Examples of important sulfoxides are alliin, a precursor to the compound that gives freshly crushed garlic its aroma, and dimethyl sulfoxide (DMSO), a common solvent.[1]
Structure and bonding

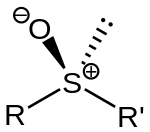
Sulfoxides feature relatively short S–O distances. In DMSO, the S–O distance is 1.531 Å. The sulfur center is pyramidal; the sum of the angles at sulfur is about 306°.[3] Sulfoxides are generally represented with the structural formula R−S(=O)−R', where R and R' are organic groups. The bond between the sulfur and oxygen atoms is intermediate of a dative bond and a polarized double bond.[4] The double-bond resonance form implies 10 electrons around sulfur (10-S-3 in N-X-L notation). The double-bond character of the S−O bond may be accounted for by donation of electron density into C−S antibonding orbitals ("no-bond" resonance forms in valence-bond language). Nevertheless, due to its simplicity and lack of ambiguity, the IUPAC recommends use of the expanded octet double-bond structure to depict sulfoxides, rather than the dipolar structure or structures that invoke "no-bond" resonance contributors.[5] The S–O interaction has an electrostatic aspect, resulting in significant dipolar character, with negative charge centered on oxygen.
Chirality
Me.svg.png.webp)
A lone pair of electrons resides on the sulfur atom, giving it tetrahedral electron-pair geometry and trigonal pyramidal shape (steric number 4 with one lone pair; see VSEPR theory). When the two organic residues are dissimilar, the sulfur is a chiral center, for example, in methyl phenyl sulfoxide. The energy barrier required to invert this stereocenter is sufficiently high that sulfoxides are optically stable near room temperature. That is, the rate of racemization is slow at room temperature. The enthalpy of activation for racemization is in the range 35 - 42 kcal/mol and the corresponding entropy of activation is -8 - +4 cal/mol-K. The barriers are lower for allylic and benzylic substituents.[6]
Preparation
Sulfoxides are typically prepared by oxidation of sulfides, sometimes referred to as sulfoxidation.[7] hydrogen peroxide is a typical oxidant, but periodate has also been used.[8] In these oxidations, care is required to avoid over oxidation to form the sulfone. For example, dimethyl sulfide is oxidized to dimethyl sulfoxide and then further to dimethyl sulfone. Unsymmetrical sulfides are prochiral, thus their oxidation gives chiral sulfoxides. This process can be performed enantioselectively.[9][10]
Aryl sulfoxides
In addition to the oxidation routes, diaryl sulfoxides can be prepared by two Friedel–Crafts arylations of sulfur dioxide using an acid catalyst:
- 2 ArH + SO2 → Ar2SO + H2O
Both aryl sulfinyl chlorides and diaryl sulfoxides can be also prepared from arenes through reaction with thionyl chloride in the presence of Lewis acid catalysts such as BiCl3, Bi(OTf)3, LiClO4, or NaClO4.[11][12]
Reactions
Deoxygenation and oxygenation
Sulfoxides undergo deoxygenation to give sulfides. Typically metal complexes are used to catalyze the reaction, using hydrosilanes as the stoichiometric reductant.[13] The deoxygenation of dimethylsulfoxide is catalyzed by DMSO reductase, a molybdoenzyme:[14]
- OSMe2 + 2 e− + 2 H+ → SMe2 + H2O
Acid-base reactions
The α-CH groups of alkyl sulfoxides are susceptible to deprotonation by strong bases, such as sodium hydride:[15]
- CH3S(O)CH3 + NaH → CH3S(O)CH2Na + H2
In the Pummerer rearrangement, alkyl sulfoxides react with acetic anhydride to give migration of the oxygen from sulfur to the adjacent carbon as an acetate ester. The first step of the reaction sequence involves the sulfoxide oxygen acting as a nucleophile:

Elimination reactions
Sulfoxide undergo thermal elimination via an Ei mechanism to yield vinyl alkenes and sulfenic acids.[16][17]
- CH3S(O)CH2CH2R → CH3SOH + CH2=CHR
The acids are powerful antioxidants, but lack long-term stability.[18] Some parent sulfoxides are therefore marketed as antioxidant polymer stabilisers.[19] Structures based on thiodipropionate esters are popular.[20] The reverse reaction is possible.
Coordination chemistry
ruthenium(II)-from-xtal-2008-3D-balls.png.webp)
Sulfoxides, especially DMSO, form coordination complexes with transition metals. Depending on the hard-soft properties of the metal, the sulfoxide binds through either the sulfur or the oxygen atom. The latter is particularly common.[21]
Applications and occurrence

DMSO is a widely used solvent.
The sulfoxide functional group occurs in several drugs. Notable is esomeprazole, the optically pure form of the proton-pump inhibitor omeprazole. Another commercially important sulfoxides include armodafinil.
Methionine sulfoxide forms from the amino acid methionine and its accumulation is associated with aging. The enzyme DMSO reductase catalyzes the interconversion of DMSO and dimethylsulfide.
Naturally-occurring chiral sulfoxides include alliin and ajoene.
Further reading
- Gama Á, Flores-López LZ, Aguirre G, Parra-Hake M, Hellberg LH, Somanathan R (2003). "Oxidation of sulfides to chiral sulfoxides using Schiff base-vanadium (IV) complexes". Arkivoc. 2003 (11): 4–15. doi:10.3998/ark.5550190.0004.b02. hdl:2027/spo.5550190.0004.b02.
References
- ↑ Patai S, Rappoport Z, eds. (1995). Syntheses of Sulphones, Sulphoxides and Cyclic Sulphides. John Wiley & Sons. doi:10.1002/9780470666357. ISBN 9780470666357.
- ↑ Yanagisawa S, Itami K (2011). "Palladium/2,2′-bipyridyl/Ag2CO3 catalyst for C–H bond arylation of heteroarenes with haloarenes". Tetrahedron. 67 (24): 4425–4430. doi:10.1016/j.tet.2011.03.093.
- ↑ Thomas R, Shoemaker CB, Eriks K (1966). "The Molecular and Crystal Structure of Dimethyl Sulfoxide, (H3C)2SO". Acta Crystallogr. 21: 12–20. doi:10.1107/S0365110X66002263..
- ↑ Cunningham TP, Cooper DL, Gerratt J, Karadakov PB, Raimondi M (1997). "Chemical bonding in oxofluorides of hypercoordinate sulfur". Journal of the Chemical Society, Faraday Transactions. 93 (13): 2247–2254. doi:10.1039/A700708F.
- ↑ Brecher J (2008). "Graphical representation standards for chemical structure diagrams" (PDF). Pure and Applied Chemistry. 80: 277–410 (on p. 389). doi:10.1351/pac200880020277. hdl:10092/2052. S2CID 98211634.
- ↑ Fernández I, Khiar N (September 2003). "Recent developments in the synthesis and utilization of chiral sulfoxides". Chemical Reviews. 103 (9): 3651–705. doi:10.1021/cr990372u. PMID 12964880.
- ↑ Roy K (2002). "Sulfones and Sulfoxides". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a25_487. ISBN 978-3527306732.
- ↑ Johnson CR, Keiser JE (1966). "Methyl Phenyl Sulfoxide". Org. Syntheses. 46: 78. doi:10.15227/orgsyn.046.0078.
- ↑ Kagan HB, Chellappan SK, Lattanzi A (2015). "(R)-(+)-Phenyl methyl sulfoxide". E-EROS Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rn00456. ISBN 978-0471936237.
- ↑ Holland, Herbert Leslie (1988). "Chiral Sulfoxidation by Biotransformation of Organic Sulfides". Chemical Reviews. 88 (3): 473–485. doi:10.1021/cr00085a002.
- ↑ Peyronneau M, Roques N, Mazières S, Le Roux C (2003). "Catalytic Lewis Acid Activation of Thionyl Chloride: Application to the Synthesis of ArylSulfinyl Chlorides Catalyzed by Bismuth(III) Salts". Synlett (5): 0631–0634. doi:10.1055/s-2003-38358.
- ↑ Bandgar BP, Makone SS (2004). "Lithium/Sodium Perchlorate Catalyzed Synthesis of Symmetrical Diaryl Sulfoxides". Synth. Commun. 34 (4): 743–750. doi:10.1081/SCC-120027723. S2CID 96348273.
- ↑ Shiri L, Kazemi M (2017). "Deoxygenation of Sulfoxides". Res. Chem. Intermed. 43: 6007–6041. doi:10.1016/j.ccr.2014.09.008.
- ↑ Sousa SC, Fernandes AC (2015). "Efficient deoxygenation methodologies catalyzed by oxo-molybdenum and oxo-rhenium complexes". Coord. Chem. Rev. 284: 67–92. doi:10.1007/s11164-017-2976-6. S2CID 102494853.
- ↑ Iwai I, Ide J (1988). "2,3-Diphenyl-1,3-Butadiene". Organic Syntheses.; Collective Volume, vol. 6, p. 531
- ↑ Michael Carrasco, Robert J. Jones, Scott Kamel, H. Rapoport, Thien Truong (1992). "N-(Benzyloxycarbonyl)-L-Vinylglycine Methyl Ester". Organic Syntheses. 70: 29. doi:10.15227/orgsyn.070.0029.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Cubbage, Jerry W.; Guo, Yushen; McCulla, Ryan D.; Jenks, William S. (1 December 2001). "Thermolysis of Alkyl Sulfoxides and Derivatives: A Comparison of Experiment and Theory". The Journal of Organic Chemistry. 66 (26): 8722–8736. doi:10.1021/jo0160625. PMID 11749600.
- ↑ Koelewijn, P.; Berger, H. (2 September 2010). "Mechanism of the antioxidant action of dialkyl sulfoxides". Recueil des Travaux Chimiques des Pays-Bas. 91 (11): 1275–1286. doi:10.1002/recl.19720911102.
- ↑ Kröhnke, C. (2016). "Polymer Stabilization". Reference Module in Materials Science and Materials Engineering. doi:10.1016/B978-0-12-803581-8.01487-9. ISBN 978-0-12-803581-8.
- ↑ Armstrong, C.; Plant, M.A.; Scott, G. (February 1975). "Mechanisms of antioxidant action: the nature of the redox behaviour of thiodipropionate esters in polypropylene". European Polymer Journal. 11 (2): 161–167. doi:10.1016/0014-3057(75)90141-X.
- ↑ Calligaris M (2004). "Structure and Bonding in Metal Sulfoxide Complexes: an Update". Coordination Chemistry Reviews. 248 (3–4): 351–375. doi:10.1016/j.ccr.2004.02.005.