 | ||||||||||||||||||||||||||||||||||||
| Sulfur | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alternative name | Sulphur (British spelling) | |||||||||||||||||||||||||||||||||||
| Allotropes | see Allotropes of sulfur | |||||||||||||||||||||||||||||||||||
| Appearance | Lemon yellow sintered microcrystals | |||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(S) | ||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||
| Sulfur in the periodic table | ||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 16 | |||||||||||||||||||||||||||||||||||
| Group | group 16 (chalcogens) | |||||||||||||||||||||||||||||||||||
| Period | period 3 | |||||||||||||||||||||||||||||||||||
| Block | p-block | |||||||||||||||||||||||||||||||||||
| Electron configuration | [Ne] 3s2 3p4 | |||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 6 | |||||||||||||||||||||||||||||||||||
| Physical properties | ||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | |||||||||||||||||||||||||||||||||||
| Melting point | 388.36 K (115.21 °C, 239.38 °F) | |||||||||||||||||||||||||||||||||||
| Boiling point | 717.8 K (444.6 °C, 832.3 °F) | |||||||||||||||||||||||||||||||||||
| Density (near r.t.) | alpha: 2.07 g/cm3 beta: 1.96 g/cm3 gamma: 1.92 g/cm3 | |||||||||||||||||||||||||||||||||||
| when liquid (at m.p.) | 1.819 g/cm3 | |||||||||||||||||||||||||||||||||||
| Critical point | 1314 K, 20.7 MPa | |||||||||||||||||||||||||||||||||||
| Heat of fusion | mono: 1.727 kJ/mol | |||||||||||||||||||||||||||||||||||
| Heat of vaporization | mono: 45 kJ/mol | |||||||||||||||||||||||||||||||||||
| Molar heat capacity | 22.75 J/(mol·K) | |||||||||||||||||||||||||||||||||||
Vapor pressure
| ||||||||||||||||||||||||||||||||||||
| Atomic properties | ||||||||||||||||||||||||||||||||||||
| Oxidation states | −2, −1, 0, +1, +2, +3, +4, +5, +6 (a strongly acidic oxide) | |||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 2.58 | |||||||||||||||||||||||||||||||||||
| Ionization energies |
| |||||||||||||||||||||||||||||||||||
| Covalent radius | 105±3 pm | |||||||||||||||||||||||||||||||||||
| Van der Waals radius | 180 pm | |||||||||||||||||||||||||||||||||||
| Other properties | ||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | |||||||||||||||||||||||||||||||||||
| Crystal structure | orthorhombic | |||||||||||||||||||||||||||||||||||
| Thermal conductivity | 0.205 W/(m⋅K) (amorphous) | |||||||||||||||||||||||||||||||||||
| Electrical resistivity | 2×1015 Ω⋅m (at 20 °C) (amorphous) | |||||||||||||||||||||||||||||||||||
| Magnetic ordering | diamagnetic[2] | |||||||||||||||||||||||||||||||||||
| Molar magnetic susceptibility | (α) −15.5×10−6 cm3/mol (298 K)[3] | |||||||||||||||||||||||||||||||||||
| Bulk modulus | 7.7 GPa | |||||||||||||||||||||||||||||||||||
| Mohs hardness | 2.0 | |||||||||||||||||||||||||||||||||||
| CAS Number | 7704-34-9 | |||||||||||||||||||||||||||||||||||
| History | ||||||||||||||||||||||||||||||||||||
| Discovery | before 2000 BCE[4] | |||||||||||||||||||||||||||||||||||
| Recognized as an element by | Antoine Lavoisier (1777) | |||||||||||||||||||||||||||||||||||
| Isotopes of sulfur | ||||||||||||||||||||||||||||||||||||
34S abundances vary greatly (between 3.96 and 4.77 percent) in natural samples. | ||||||||||||||||||||||||||||||||||||
Sulfur (also spelled sulphur in British English) is a chemical element; it has symbol S and atomic number 16. It is abundant, multivalent and nonmetallic. Under normal conditions, sulfur atoms form cyclic octatomic molecules with the chemical formula S8. Elemental sulfur is a bright yellow, crystalline solid at room temperature.
Sulfur is the tenth most abundant element by mass in the universe and the fifth most abundant on Earth. Though sometimes found in pure, native form, sulfur on Earth usually occurs as sulfide and sulfate minerals. Being abundant in native form, sulfur was known in ancient times, being mentioned for its uses in ancient India, ancient Greece, China, and ancient Egypt. Historically and in literature sulfur is also called brimstone,[5] which means "burning stone".[6] Today, almost all elemental sulfur is produced as a byproduct of removing sulfur-containing contaminants from natural gas and petroleum.[7][8] The greatest commercial use of the element is the production of sulfuric acid for sulfate and phosphate fertilizers, and other chemical processes. Sulfur is used in matches, insecticides, and fungicides. Many sulfur compounds are odoriferous, and the smells of odorized natural gas, skunk scent, bad breath, grapefruit, and garlic are due to organosulfur compounds. Hydrogen sulfide gives the characteristic odor to rotting eggs and other biological processes.
Sulfur is an essential element for all life, almost always in the form of organosulfur compounds or metal sulfides. Amino acids (two proteinogenic: cysteine and methionine, and many other non-coded: cystine, taurine, etc.) and two vitamins (biotin and thiamine) are organosulfur compounds crucial for life. Many cofactors also contain sulfur, including glutathione, and iron–sulfur proteins. Disulfides, S–S bonds, confer mechanical strength and insolubility of the (among others) protein keratin, found in outer skin, hair, and feathers. Sulfur is one of the core chemical elements needed for biochemical functioning and is an elemental macronutrient for all living organisms.
Characteristics
Physical properties

Sulfur forms several polyatomic molecules. The best-known allotrope is octasulfur, cyclo-S8. The point group of cyclo-S8 is D4d and its dipole moment is 0 D.[9] Octasulfur is a soft, bright-yellow solid that is odorless, but impure samples have an odor similar to that of matches.[10] It melts at 115.21 °C (239.38 °F), boils at 444.6 °C (832.3 °F)[5] and sublimes more or less between 20 °C (68 °F) and 50 °C (122 °F).[11] At 95.2 °C (203.4 °F), below its melting temperature, cyclo-octasulfur changes from α-octasulfur to the β-polymorph.[12] The structure of the S8 ring is virtually unchanged by this phase change, which affects the intermolecular interactions. Between its melting and boiling temperatures, octasulfur changes its allotrope again, turning from β-octasulfur to γ-sulfur, again accompanied by a lower density but increased viscosity due to the formation of polymers.[12] At higher temperatures, the viscosity decreases as depolymerization occurs. Molten sulfur assumes a dark red color above 200 °C (392 °F). The density of sulfur is about 2 g/cm3, depending on the allotrope; all of the stable allotropes are excellent electrical insulators.
Sulfur is insoluble in water but soluble in carbon disulfide and, to a lesser extent, in other nonpolar organic solvents, such as benzene and toluene.
Chemical properties
Under normal conditions, sulfur hydrolyzes very slowly to mainly form hydrogen sulfide and sulfuric acid:
8 + 4 H
2O → 3 H
2S + H
2SO
4
The reaction involves adsorption of protons onto S
8 clusters, followed by disproportionation into the reaction products.[13]
The second, fourth and sixth ionization energies of sulfur are 2252 kJ/mol, 4556 kJ/mol and 8495.8 kJ/mol, respectively. A composition of products of sulfur's reactions with oxidants (and its oxidation state) depends on that whether releasing out of a reaction energy overcomes these thresholds. Applying catalysts and / or supply of outer energy may vary sulfur's oxidation state and a composition of reaction products. While reaction between sulfur and oxygen at normal conditions gives sulfur dioxide (oxidation state +4), formation of sulfur trioxide (oxidation state +6) requires temperature 400–600 °C (750–1,100 °F) and presence of a catalyst.
In reactions with elements of lesser electronegativity, it reacts as an oxidant and forms sulfides, where it has oxidation state −2.
Sulfur reacts with nearly all other elements with the exception of the noble gases, even with the notoriously unreactive metal iridium (yielding iridium disulfide).[14] Some of those reactions need elevated temperatures.[15]
Allotropes
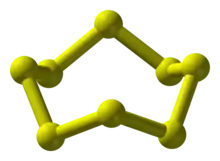
Sulfur forms over 30 solid allotropes, more than any other element.[16] Besides S8, several other rings are known.[17] Removing one atom from the crown gives S7, which is of a deeper yellow than S8. HPLC analysis of "elemental sulfur" reveals an equilibrium mixture of mainly S8, but with S7 and small amounts of S6.[18] Larger rings have been prepared, including S12 and S18.[19][20]
Amorphous or "plastic" sulfur is produced by rapid cooling of molten sulfur—for example, by pouring it into cold water. X-ray crystallography studies show that the amorphous form may have a helical structure with eight atoms per turn. The long coiled polymeric molecules make the brownish substance elastic, and in bulk this form has the feel of crude rubber. This form is metastable at room temperature and gradually reverts to the crystalline molecular allotrope, which is no longer elastic. This process happens within a matter of hours to days, but can be rapidly catalyzed.
Isotopes
Sulfur has 23 known isotopes, four of which are stable: 32S (94.99%±0.26%), 33S (0.75%±0.02%), 34S (4.25%±0.24%), and 36S (0.01%±0.01%).[21][22] Other than 35S, with a half-life of 87 days, the radioactive isotopes of sulfur have half-lives less than 3 hours.
The preponderance of 32S is explained by its production in the so-called alpha-process (one of the main classes of nuclear fusion reactions) in exploding stars. Other stable sulfur isotopes are produced in the bypass processes related with 34Ar, and their composition depends on a type of a stellar explosion. For example, proportionally more 33S comes from novae than from supernovae.[23]
On the planet Earth the sulfur isotopic composition was determined by the Sun. Though it is assumed that the distribution of different sulfur isotopes should be more or less equal, it has been found that proportions of two most abundant sulfur isotopes 32S and 34S varies in different samples. Assaying of these isotopes ratio (δ34S) in the samples allows to make suggestions about their chemical history, and with support of other methods, it allows to age-date the samples, estimate temperature of equilibrium between ore and water, determine pH and oxygen fugacity, identify the activity of sulfate-reducing bacteria in the time of formation of the sample, or suggest the main sources of sulfur in ecosystems.[24] However, there are ongoing discussions about what is the real reason of the δ34S shifts, biological activity or postdeposital alteration.[25]
For example, when sulfide minerals are precipitated, isotopic equilibration among solids and liquid may cause small differences in the δ34S values of co-genetic minerals. The differences between minerals can be used to estimate the temperature of equilibration. The δ13C and δ34S of coexisting carbonate minerals and sulfides can be used to determine the pH and oxygen fugacity of the ore-bearing fluid during ore formation.
Scientists measure the sulfur isotopes of minerals in rocks and sediments to study the redox conditions in the oceans in the past. Sulfate-reducing bacteria in marine sediment fractionate sulfur isotopes as they take in sulfate and produce sulfide. Prior to 2010s, it was thought that sulfate reduction could fractionate sulfur isotopes up to 46 permil[26] and fractionation larger than 46 permil recorded in sediments must be due to disproportionation of sulfur compounds in the sediment. This view has changed since the 2010s as experiments show that sulfate-reducing bacteria can fractionate to 66 permil.[27] As substrates for disproportionation are limited by the product of sulfate reduction, the isotopic effect of disproportionation should be less than 16 permil in most sedimentary settings.[28]
In most forest ecosystems, sulfate is derived mostly from the atmosphere; weathering of ore minerals and evaporites contribute some sulfur. Sulfur with a distinctive isotopic composition has been used to identify pollution sources, and enriched sulfur has been added as a tracer in hydrologic studies. Differences in the natural abundances can be used in systems where there is sufficient variation in the 34S of ecosystem components. Rocky Mountain lakes thought to be dominated by atmospheric sources of sulfate have been found to have measurably different 34S values than lakes believed to be dominated by watershed sources of sulfate.
The radioactive 35S is formed in cosmic ray spallation of the atmospheric 40Ar. This fact may be used for proving the presence of recent (not more than 1 year) atmospheric sediments in various things. This isotope may be obtained artificially by different ways. In practice, the reaction 35Cl + n → 35S + p is used by irradiating potassium chloride with neutrons.[29] The isotope 35S is used in various sulfur-containing compounds as a radioactive tracer for many biological studies, for example, the Hershey-Chase experiment.
Because of the weak beta activity of 35S, its compounds are relatively safe as long as they are not ingested or absorbed by the body.[30]
Natural occurrence




32S is created inside massive stars, at a depth where the temperature exceeds 2.5×109 K, by the fusion of one nucleus of silicon plus one nucleus of helium.[31] As this nuclear reaction is part of the alpha process that produces elements in abundance, sulfur is the 10th most common element in the universe.
Sulfur, usually as sulfide, is present in many types of meteorites. Ordinary chondrites contain on average 2.1% sulfur, and carbonaceous chondrites may contain as much as 6.6%. It is normally present as troilite (FeS), but there are exceptions, with carbonaceous chondrites containing free sulfur, sulfates and other sulfur compounds.[32] The distinctive colors of Jupiter's volcanic moon Io are attributed to various forms of molten, solid, and gaseous sulfur.[33]
It is the fifth most common element by mass in the Earth. Elemental sulfur can be found near hot springs and volcanic regions in many parts of the world, especially along the Pacific Ring of Fire; such volcanic deposits are currently mined in Indonesia, Chile, and Japan. These deposits are polycrystalline, with the largest documented single crystal measuring 22×16×11 cm.[34] Historically, Sicily was a major source of sulfur in the Industrial Revolution.[35] Lakes of molten sulfur up to ~200 m in diameter have been found on the sea floor, associated with submarine volcanoes, at depths where the boiling point of water is higher than the melting point of sulfur.[36]
Native sulfur is synthesised by anaerobic bacteria acting on sulfate minerals such as gypsum in salt domes.[37][38] Significant deposits in salt domes occur along the coast of the Gulf of Mexico, and in evaporites in eastern Europe and western Asia. Native sulfur may be produced by geological processes alone. Fossil-based sulfur deposits from salt domes were once the basis for commercial production in the United States, Russia, Turkmenistan, and Ukraine.[39] Currently, commercial production is still carried out in the Osiek mine in Poland. Such sources are now of secondary commercial importance, and most are no longer worked.
Common naturally occurring sulfur compounds include the sulfide minerals, such as pyrite (iron sulfide), cinnabar (mercury sulfide), galena (lead sulfide), sphalerite (zinc sulfide), and stibnite (antimony sulfide); and the sulfate minerals, such as gypsum (calcium sulfate), alunite (potassium aluminium sulfate), and barite (barium sulfate). On Earth, just as upon Jupiter's moon Io, elemental sulfur occurs naturally in volcanic emissions, including emissions from hydrothermal vents.
The main industrial source of sulfur is now petroleum and natural gas.[7]
Compounds
Common oxidation states of sulfur range from −2 to +6. Sulfur forms stable compounds with all elements except the noble gases.
Electron transfer reactions

Sulfur polycations, S82+, S42+ and S162+ are produced when sulfur is reacted with oxidising agents in a strongly acidic solution.[40] The colored solutions produced by dissolving sulfur in oleum were first reported as early as 1804 by C.F. Bucholz, but the cause of the color and the structure of the polycations involved was only determined in the late 1960s. S82+ is deep blue, S42+ is yellow and S162+ is red.[12]
Reduction of sulfur gives various polysulfides with the formula Sx2−, many of which have been obtained in crystalline form. Illustrative is the production of sodium tetrasulfide:
Some of these dianions dissociate to give radical anions, such as S3− gives the blue color of the rock lapis lazuli.
This reaction highlights a distinctive property of sulfur: its ability to catenate (bind to itself by formation of chains). Protonation of these polysulfide anions produces the polysulfanes, H2Sx, where x = 2, 3, and 4.[42] Ultimately, reduction of sulfur produces sulfide salts:
The interconversion of these species is exploited in the sodium–sulfur battery.
Hydrogenation
Treatment of sulfur with hydrogen gives hydrogen sulfide. When dissolved in water, hydrogen sulfide is mildly acidic:[5]
Hydrogen sulfide gas and the hydrosulfide anion are extremely toxic to mammals, due to their inhibition of the oxygen-carrying capacity of hemoglobin and certain cytochromes in a manner analogous to cyanide and azide (see below, under precautions).
Combustion
The two principal sulfur oxides are obtained by burning sulfur:
Many other sulfur oxides are observed including the sulfur-rich oxides include sulfur monoxide, disulfur monoxide, disulfur dioxides, and higher oxides containing peroxo groups.
Halogenation
Sulfur reacts with fluorine to give the highly reactive sulfur tetrafluoride and the highly inert sulfur hexafluoride.[43] Whereas fluorine gives S(IV) and S(VI) compounds, chlorine gives S(II) and S(I) derivatives. Thus, sulfur dichloride, disulfur dichloride, and higher chlorosulfanes arise from the chlorination of sulfur. Sulfuryl chloride and chlorosulfuric acid are derivatives of sulfuric acid; thionyl chloride (SOCl2) is a common reagent in organic synthesis.[44] Similar to chlorine, bromine will also oxidize sulfur to form sulfur dibromide and disulfur dibromide.[44] The former is unstable and readily decomposes to the latter. Meanwhile, sulfur iodides are extremely unstable for reasons relating to their electronegativity values, and so are only known under extremely low temperatures.
Pseudohalides
Sulfur oxidizes cyanide and sulfite to give thiocyanate and thiosulfate, respectively.
Metal sulfides
Sulfur reacts with many metals. Electropositive metals give polysulfide salts. Copper, zinc, and silver are attacked by sulfur; see tarnishing. Although many metal sulfides are known, most are prepared by high temperature reactions of the elements.[45] Geoscientists also study the isotopes of metal sulfides in rocks and sediment to study environmental conditions in the Earth's past.[46]
Organic compounds
- Illustrative organosulfur compounds
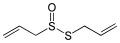 Allicin, a chemical compound in garlic
Allicin, a chemical compound in garlic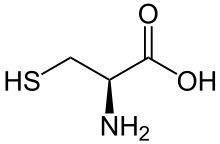 (L)-cysteine, an amino acid containing a thiol group
(L)-cysteine, an amino acid containing a thiol group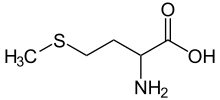 Methionine, an amino acid containing a thioether
Methionine, an amino acid containing a thioether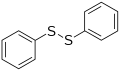 Diphenyl disulfide, a representative disulfide
Diphenyl disulfide, a representative disulfide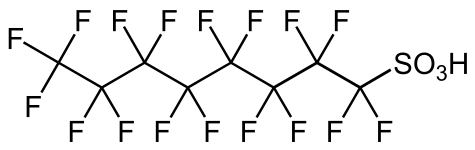 Perfluorooctanesulfonic acid, a surfactant
Perfluorooctanesulfonic acid, a surfactant Dibenzothiophene, a component of crude oil
Dibenzothiophene, a component of crude oil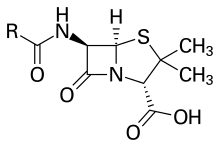 Penicillin, an antibiotic where "R" is the variable group
Penicillin, an antibiotic where "R" is the variable group
Some of the main classes of sulfur-containing organic compounds include the following:[47]
- Thiols or mercaptans (so called because they capture mercury as chelators) are the sulfur analogs of alcohols; treatment of thiols with base gives thiolate ions.
- Thioethers are the sulfur analogs of ethers.
- Sulfonium ions have three groups attached to a cationic sulfur center. Dimethylsulfoniopropionate (DMSP) is one such compound, important in the marine organic sulfur cycle.
- Sulfoxides and sulfones are thioethers with one and two oxygen atoms attached to the sulfur atom, respectively. The simplest sulfoxide, dimethyl sulfoxide, is a common solvent; a common sulfone is sulfolane.
- Sulfonic acids are used in many detergents.
Compounds with carbon–sulfur multiple bonds are uncommon, an exception being carbon disulfide, a volatile colorless liquid that is structurally similar to carbon dioxide. It is used as a reagent to make the polymer rayon and many organosulfur compounds. Unlike carbon monoxide, carbon monosulfide is stable only as an extremely dilute gas, found between solar systems.[48]
Organosulfur compounds are responsible for some of the unpleasant odors of decaying organic matter. They are widely known as the odorant in domestic natural gas, garlic odor, and skunk spray, as well as a component of bad breath odor. Not all organic sulfur compounds smell unpleasant at all concentrations: the sulfur-containing monoterpenoid grapefruit mercaptan in small concentrations is the characteristic scent of grapefruit, but has a generic thiol odor at larger concentrations. Sulfur mustard, a potent vesicant, was used in World War I as a disabling agent.[49]
Sulfur–sulfur bonds are a structural component used to stiffen rubber, similar to the disulfide bridges that rigidify proteins (see biological below). In the most common type of industrial "curing" or hardening and strengthening of natural rubber, elemental sulfur is heated with the rubber to the point that chemical reactions form disulfide bridges between isoprene units of the polymer. This process, patented in 1843, made rubber a major industrial product, especially in automobile tires. Because of the heat and sulfur, the process was named vulcanization, after the Roman god of the forge and volcanism.
History
Antiquity

Being abundantly available in native form, sulfur was known in ancient times and is referred to in the Torah (Genesis). English translations of the Christian Bible commonly referred to burning sulfur as "brimstone", giving rise to the term "fire-and-brimstone" sermons, in which listeners are reminded of the fate of eternal damnation that await the unbelieving and unrepentant. It is from this part of the Bible[50] that Hell is implied to "smell of sulfur" (likely due to its association with volcanic activity). According to the Ebers Papyrus, a sulfur ointment was used in ancient Egypt to treat granular eyelids. Sulfur was used for fumigation in preclassical Greece;[51] this is mentioned in the Odyssey.[52] Pliny the Elder discusses sulfur in book 35 of his Natural History, saying that its best-known source is the island of Melos. He mentions its use for fumigation, medicine, and bleaching cloth.[53]
A natural form of sulfur known as shiliuhuang (石硫黄) was known in China since the 6th century BC and found in Hanzhong.[54] By the 3rd century, the Chinese had discovered that sulfur could be extracted from pyrite.[54] Chinese Daoists were interested in sulfur's flammability and its reactivity with certain metals, yet its earliest practical uses were found in traditional Chinese medicine.[54] The Wujing Zongyao of 1044 AD described various formulas for Chinese black powder, which is a mixture of potassium nitrate (KNO
3), charcoal, and sulfur.[55]
.svg.png.webp)
.svg.png.webp)
Indian alchemists, practitioners of the "science of chemicals" (Sanskrit: रसशास्त्र, romanized: rasaśāstra), wrote extensively about the use of sulfur in alchemical operations with mercury, from the eighth century AD onwards.[57] In the rasaśāstra tradition, sulfur is called "the smelly" (गन्धक, gandhaka).
Early European alchemists gave sulfur a unique alchemical symbol, a triangle atop a cross (🜍). (This is sometimes confused with the astronomical crossed-spear symbol ⚴ for 2 Pallas.) The variation known as brimstone has a symbol combining a two-barred cross atop a lemniscate (🜏). In traditional skin treatment, elemental sulfur was used (mainly in creams) to alleviate such conditions as scabies, ringworm, psoriasis, eczema, and acne. The mechanism of action is unknown—though elemental sulfur does oxidize slowly to sulfurous acid, which is (through the action of sulfite) a mild reducing and antibacterial agent.[58][59][60]
Modern times

Sulfur appears in a column of fixed (non-acidic) alkali in a chemical table of 1718.[62] Antoine Lavoisier used sulfur in combustion experiments, writing of some of these in 1777.[63]
Sulfur deposits in Sicily were the dominant source for more than a century. By the late 18th century, about 2,000 tonnes per year of sulfur were imported into Marseille, France, for the production of sulfuric acid for use in the Leblanc process. In industrializing Britain, with the repeal of tariffs on salt in 1824, demand for sulfur from Sicily surged upward. The increasing British control and exploitation of the mining, refining, and transportation of the sulfur, coupled with the failure of this lucrative export to transform Sicily's backward and impoverished economy, led to the Sulfur Crisis of 1840, when King Ferdinand II gave a monopoly of the sulfur industry to a French firm, violating an earlier 1816 trade agreement with Britain. A peaceful solution was eventually negotiated by France.[64][65]
In 1867, elemental sulfur was discovered in underground deposits in Louisiana and Texas. The highly successful Frasch process was developed to extract this resource.[66]
In the late 18th century, furniture makers used molten sulfur to produce decorative inlays.[67] Molten sulfur is sometimes still used for setting steel bolts into drilled concrete holes where high shock resistance is desired for floor-mounted equipment attachment points. Pure powdered sulfur was used as a medicinal tonic and laxative.[39]
With the advent of the contact process, the majority of sulfur today is used to make sulfuric acid for a wide range of uses, particularly fertilizer.[68]
In recent times, the main source of sulfur has become petroleum and natural gas. This is due to the requirement to remove sulfur from fuels in order to prevent acid rain, and has resulted in a surplus of sulfur.[7]
Spelling and etymology
Sulfur is derived from the Latin word sulpur, which was Hellenized to sulphur in the erroneous belief that the Latin word came from Greek. This spelling was later reinterpreted as representing an /f/ sound and resulted in the spelling sulfur, which appears in Latin toward the end of the Classical period. The true Ancient Greek word for sulfur, θεῖον, theîon (from earlier θέειον, théeion), is the source of the international chemical prefix thio-. The Modern Standard Greek word for sulfur is θείο, theío.
In 12th-century Anglo-French, it was sulfre. In the 14th century, the erroneously Hellenized Latin -ph- was restored in Middle English sulphre. By the 15th century, both full Latin spelling variants sulfur and sulphur became common in English. The parallel f~ph spellings continued in Britain until the 19th century, when the word was standardized as sulphur.[69] On the other hand, sulfur was the form chosen in the United States, whereas Canada uses both.
The IUPAC adopted the spelling sulfur in 1990 or 1971, depending on the source cited,[70] as did the Nomenclature Committee of the Royal Society of Chemistry in 1992, restoring the spelling sulfur to Britain.[71] Oxford Dictionaries note that "in chemistry and other technical uses ... the -f- spelling is now the standard form for this and related words in British as well as US contexts, and is increasingly used in general contexts as well."[72]
Production


Sulfur may be found by itself and historically was usually obtained in this form; pyrite has also been a source of sulfur.[73] In volcanic regions in Sicily, in ancient times, it was found on the surface of the Earth, and the "Sicilian process" was used: sulfur deposits were piled and stacked in brick kilns built on sloping hillsides, with airspaces between them. Then, some sulfur was pulverized, spread over the stacked ore and ignited, causing the free sulfur to melt down the hills. Eventually the surface-borne deposits played out, and miners excavated veins that ultimately dotted the Sicilian landscape with labyrinthine mines. Mining was unmechanized and labor-intensive, with pickmen freeing the ore from the rock, and mine-boys or carusi carrying baskets of ore to the surface, often through a mile or more of tunnels. Once the ore was at the surface, it was reduced and extracted in smelting ovens. The conditions in Sicilian sulfur mines were horrific, prompting Booker T. Washington to write "I am not prepared just now to say to what extent I believe in a physical hell in the next world, but a sulfur mine in Sicily is about the nearest thing to hell that I expect to see in this life."[74] Sulfur is still mined from surface deposits in poorer nations with volcanoes, such as Indonesia, and worker conditions have not improved much since Booker T. Washington's days.[75]
Elemental sulfur was extracted from salt domes (in which it sometimes occurs in nearly pure form) until the late 20th century. Sulfur is now produced as a side product of other industrial processes such as in oil refining, in which sulfur is undesired. As a mineral, native sulfur under salt domes is thought to be a fossil mineral resource, produced by the action of anaerobic bacteria on sulfate deposits. It was removed from such salt-dome mines mainly by the Frasch process.[39] In this method, superheated water was pumped into a native sulfur deposit to melt the sulfur, and then compressed air returned the 99.5% pure melted product to the surface. Throughout the 20th century this procedure produced elemental sulfur that required no further purification. Due to a limited number of such sulfur deposits and the high cost of working them, this process for mining sulfur has not been employed in a major way anywhere in the world since 2002.[76][77]

Today, sulfur is produced from petroleum, natural gas, and related fossil resources, from which it is obtained mainly as hydrogen sulfide.[7] Organosulfur compounds, undesirable impurities in petroleum, may be upgraded by subjecting them to hydrodesulfurization, which cleaves the C–S bonds:[76][77]
The resulting hydrogen sulfide from this process, and also as it occurs in natural gas, is converted into elemental sulfur by the Claus process. This process entails oxidation of some hydrogen sulfide to sulfur dioxide and then the comproportionation of the two:[76][77]
Owing to the high sulfur content of the Athabasca Oil Sands, stockpiles of elemental sulfur from this process now exist throughout Alberta, Canada.[78] Another way of storing sulfur is as a binder for concrete, the resulting product having many desirable properties (see sulfur concrete).[79]
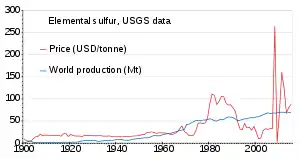
The world production of sulfur in 2011 amounted to 69 million tonnes (Mt), with more than 15 countries contributing more than 1 Mt each. Countries producing more than 5 Mt are China (9.6), the United States (8.8), Canada (7.1) and Russia (7.1).[80] Production has been slowly increasing from 1900 to 2010; the price was unstable in the 1980s and around 2010.[81]
Applications
Sulfuric acid
Elemental sulfur is used mainly as a precursor to other chemicals. Approximately 85% (1989) is converted to sulfuric acid (H2SO4):

In 2010, the United States produced more sulfuric acid than any other inorganic industrial chemical.[81] The principal use for the acid is the extraction of phosphate ores for the production of fertilizer manufacturing. Other applications of sulfuric acid include oil refining, wastewater processing, and mineral extraction.[39]
Other important sulfur chemistry
Sulfur reacts directly with methane to give carbon disulfide, which is used to manufacture cellophane and rayon.[39] One of the uses of elemental sulfur is in vulcanization of rubber, where polysulfide chains crosslink organic polymers. Large quantities of sulfites are used to bleach paper and to preserve dried fruit. Many surfactants and detergents (e.g. sodium lauryl sulfate) are sulfate derivatives. Calcium sulfate, gypsum (CaSO4·2H2O) is mined on the scale of 100 million tonnes each year for use in Portland cement and fertilizers.
When silver-based photography was widespread, sodium and ammonium thiosulfate were widely used as "fixing agents". Sulfur is a component of gunpowder ("black powder").
Fertilizer
Amino acids synthesized by living organisms such as methionine and cysteine contain organosulfur groups (thioester and thiol respectively). The antioxidant glutathione protecting many living organisms against free radicals and oxidative stress also contains organic sulfur. Some crops such as onion and garlic also produce different organosulfur compounds such as syn-propanethial-S-oxide responsible of lacrymal irritation (onions), or diallyl disulfide and allicin (garlic). Sulfates, commonly found in soils and groundwaters are often a sufficient natural source of sulfur for plants and bacteria. Atmospheric deposition of sulfur dioxide (SO2) is also a common artificial source (coal combustion) of sulfur for the soils. Under normal circumstances, in most agricultural soils, sulfur is not a limiting nutrient for plants and microorganisms (see Liebig's barrel). However, in some circumstances, soils can be depleted in sulfate, e.g. if this later is leached by meteoric water (rain) or if the requirements in sulfur for some types of crops are high. This explains that sulfur is increasingly recognized and used as a component of fertilizers. The most important form of sulfur for fertilizer is calcium sulfate, commonly found in nature as the mineral gypsum (CaSO4·2H2O). Elemental sulfur is hydrophobic (not soluble in water) and cannot be used directly by plants. Elemental sulfur (ES) is sometimes mixed with bentonite to amend depleted soils for crops with high requirement in organo-sulfur. Over time, oxidation abiotic processes with atmospheric oxygen and soil bacteria can oxidize and convert elemental sulfur to soluble derivatives, which can then be used by microorganisms and plants. Sulfur improves the efficiency of other essential plant nutrients, particularly nitrogen and phosphorus.[82] Biologically produced sulfur particles are naturally hydrophilic due to a biopolymer coating and are easier to disperse over the land in a spray of diluted slurry, resulting in a faster uptake by plants.
The plants requirement for sulfur equals or exceeds the requirement for phosphorus. It is an essential nutrient for plant growth, root nodule formation of legumes, and immunity and defense systems. Sulfur deficiency has become widespread in many countries in Europe.[83][84][85] Because atmospheric inputs of sulfur continue to decrease, the deficit in the sulfur input/output is likely to increase unless sulfur fertilizers are used. Atmospheric inputs of sulfur decrease because of actions taken to limit acid rains.[86][82]
Fungicide and pesticide

Elemental sulfur is one of the oldest fungicides and pesticides. "Dusting sulfur", elemental sulfur in powdered form, is a common fungicide for grapes, strawberry, many vegetables and several other crops. It has a good efficacy against a wide range of powdery mildew diseases as well as black spot. In organic production, sulfur is the most important fungicide. It is the only fungicide used in organically farmed apple production against the main disease apple scab under colder conditions. Biosulfur (biologically produced elemental sulfur with hydrophilic characteristics) can also be used for these applications.
Standard-formulation dusting sulfur is applied to crops with a sulfur duster or from a dusting plane. Wettable sulfur is the commercial name for dusting sulfur formulated with additional ingredients to make it water miscible.[79][87] It has similar applications and is used as a fungicide against mildew and other mold-related problems with plants and soil.
Elemental sulfur powder is used as an "organic" (i.e., "green") insecticide (actually an acaricide) against ticks and mites. A common method of application is dusting the clothing or limbs with sulfur powder.
A diluted solution of lime sulfur (made by combining calcium hydroxide with elemental sulfur in water) is used as a dip for pets to destroy ringworm (fungus), mange, and other dermatoses and parasites.
Sulfur candles of almost pure sulfur were burned to fumigate structures and wine barrels, but are now considered too toxic for residences.
Pharmaceuticals
Sulfur (specifically octasulfur, S8) is used in pharmaceutical skin preparations for the treatment of acne and other conditions. It acts as a keratolytic agent and also kills bacteria, fungi, scabies mites, and other parasites.[88] Precipitated sulfur and colloidal sulfur are used, in form of lotions, creams, powders, soaps, and bath additives, for the treatment of acne vulgaris, acne rosacea, and seborrhoeic dermatitis.[89]
Many drugs contain sulfur.[90] Early examples include antibacterial sulfonamides, known as sulfa drugs. A more recent example is mucolytic acetylcysteine. Sulfur is a part of many bacterial defense molecules. Most β-lactam antibiotics, including the penicillins, cephalosporins and monobactams contain sulfur.[47]
Batteries
Due to their high energy density and the availability of sulfur, there is ongoing research in creating rechargeable lithium–sulfur batteries. Until now, carbonate electrolytes have caused failures in such batteries after a single cycle. In February 2022, researchers at Drexel University have not only created a prototypical battery that lasted 4000 recharge cycles, but also found the first monoclinic gamma sulfur that remained stable below 95 degrees Celsius.[91]
Biological role
Sulfur is an essential component of all living cells. It is the eighth most abundant element in the human body by weight,[92] about equal in abundance to potassium, and slightly greater than sodium and chlorine.[93] A 70 kg (150 lb) human body contains about 140 grams of sulfur.[94] The main dietary source of sulfur for humans is sulfur-containing amino-acids,[95] which can be found in plant and animal proteins.[96]
Transferring sulfur between inorganic and biomolecules
In the 1880s, while studying Beggiatoa (a bacterium living in a sulfur rich environment), Sergei Winogradsky found that it oxidized hydrogen sulfide (H2S) as an energy source, forming intracellular sulfur droplets. Winogradsky referred to this form of metabolism as inorgoxidation (oxidation of inorganic compounds).[97] Another contributor, who continued to study it was Selman Waksman.[98] Primitive bacteria that live around deep ocean volcanic vents oxidize hydrogen sulfide for their nutrition, as discovered by Robert Ballard.[8]
Sulfur oxidizers can use as energy sources reduced sulfur compounds, including hydrogen sulfide, elemental sulfur, sulfite, thiosulfate, and various polythionates (e.g., tetrathionate).[99] They depend on enzymes such as sulfur oxygenase and sulfite oxidase to oxidize sulfur to sulfate. Some lithotrophs can even use the energy contained in sulfur compounds to produce sugars, a process known as chemosynthesis. Some bacteria and archaea use hydrogen sulfide in place of water as the electron donor in chemosynthesis, a process similar to photosynthesis that produces sugars and uses oxygen as the electron acceptor. Sulfur-based chemosynthesis may be simplifiedly compared with photosynthesis:
There are bacteria combining these two ways of nutrition: green sulfur bacteria and purple sulfur bacteria.[100] Also sulfur-oxidizing bacteria can go into symbiosis with larger organisms, enabling the later to use hydrogen sulfide as food to be oxidized. Example: the giant tube worm.[101]
There are sulfate-reducing bacteria, that, by contrast, "breathe sulfate" instead of oxygen. They use organic compounds or molecular hydrogen as the energy source. They use sulfur as the electron acceptor, and reduce various oxidized sulfur compounds back into sulfide, often into hydrogen sulfide. They can grow on other partially oxidized sulfur compounds (e.g. thiosulfates, thionates, polysulfides, sulfites).
There are studies pointing that many deposits of native sulfur in places that were the bottom of the ancient oceans have biological origin.[102][103][104] These studies indicate that this native sulfur have been obtained through biological activity, but what is responsible for that (sulfur-oxidizing bacteria or sulfate-reducing bacteria) is still unknown for sure.
Sulfur is absorbed by plants roots from soil as sulfate and transported as a phosphate ester. Sulfate is reduced to sulfide via sulfite before it is incorporated into cysteine and other organosulfur compounds.[105]
While the plants' role in transferring sulfur to animals by food chains is more or less understood, the role of sulfur bacteria is just getting investigated.[106][107]
Protein and organic metabolites
In all forms of life, most of the sulfur is contained in two proteinogenic amino acids (cysteine and methionine), thus the element is present in all proteins that contain these amino acids, as well as in respective peptides.[108] Some of the sulfur is comprised in certain metabolites — many of which are cofactors — and sulfated polysaccharides of connective tissue (chondroitin sulfates, heparin).
Proteins, to execute their biological function, need to have specific space geometry. Formation of this geometry is performed in a process called protein folding, and is provided by intra- and inter-molecular bonds. The process has several stages. While at premier stages a polypeptide chain folds due to hydrogen bonds, at later stages folding is provided (apart from hydrogen bonds) by covalent bonds between two sulfur atoms of two cysteine residues (so called disulfide bridges) at different places of a chain (tertiary protein structure) as well as between two cysteine residues in two separated protein subunits (quaternary protein structure). Both structures easily may be seen in insulin. As the bond energy of a covalent disulfide bridge is higher than the energy of a coordinate bond or hydrophobic interaction, higher disulfide bridges content leads to higher energy needed for protein denaturation. In general disulfide bonds are necessary in proteins functioning outside cellular space, and they do not change proteins' conformation (geometry), but serve as its stabilizers.[109] Within cytoplasm cysteine residues of proteins are saved in reduced state (i.e. in -SH form) by thioredoxins.[110]
This property manifests in following examples. Lysozyme is stable enough to be applied as a drug.[111] Feathers and hair have relative strength, and consisting in them keratin is considered indigestible by most organisms. However, there are fungi and bacteria containing keratinase, and are able to destruct keratin.
Many important cellular enzymes use prosthetic groups ending with -SH moieties to handle reactions involving acyl-containing biochemicals: two common examples from basic metabolism are coenzyme A and alpha-lipoic acid.[112] Cysteine-related metabolites homocysteine and taurine are other sulfur-containing amino acids that are similar in structure, but not coded by DNA, and are not part of the primary structure of proteins, take part in various locations of mammalian physiology.[113][114] Two of the 13 classical vitamins, biotin and thiamine, contain sulfur, and serve as cofactors to several enzymes.[115][116]
In intracellular chemistry, sulfur operates as a carrier of reducing hydrogen and its electrons for cellular repair of oxidation. Reduced glutathione, a sulfur-containing tripeptide, is a reducing agent through its sulfhydryl (-SH) moiety derived from cysteine.
Methanogenesis, the route to most of the world's methane, is a multistep biochemical transformation of carbon dioxide. This conversion requires several organosulfur cofactors. These include coenzyme M, CH3SCH2CH2SO3−, the immediate precursor to methane.[117]
Metalloproteins and inorganic cofactors
Metalloproteins — in which the active site is a transition metal ion (or metal-sulfide cluster) often coordinated by sulfur atoms of cysteine residues[118] — are essential components of enzymes involved in electron transfer processes. Examples include plastocyanin (Cu2+) and nitrous oxide reductase (Cu–S). The function of these enzymes is dependent on the fact that the transition metal ion can undergo redox reactions. Other examples include many zinc proteins,[119] as well as iron–sulfur clusters. Most pervasive are the ferrodoxins, which serve as electron shuttles in cells. In bacteria, the important nitrogenase enzymes contain an Fe–Mo–S cluster and is a catalyst that performs the important function of nitrogen fixation, converting atmospheric nitrogen to ammonia that can be used by microorganisms and plants to make proteins, DNA, RNA, alkaloids, and the other organic nitrogen compounds necessary for life.[120]
 Easiness of electron flow in a cluster provides catalytic effect of a respective enzyme.
Easiness of electron flow in a cluster provides catalytic effect of a respective enzyme.
Deficiency
In humans methionine is an essential amino acid; cysteine is conditionally essential and may be synthesized from non-essential serine (sulfur donor would be methionine in this case). Dietary deficiency rarely happens in common conditions. Artificial methionine deficiency is attempted to apply in cancer treatment,[121] but the method is still potentially dangerous.[122]
Isolated sulfite oxidase deficiency is a rare, fatal genetic disease preventing production of sulfite oxidase, needed to metabolize sulfites to sulfates.[123]
Precautions
| Hazards | |
|---|---|
| GHS labelling: | |
  | |
| Warning | |
| H315[124] | |
| NFPA 704 (fire diamond) | |


Though elemental sulfur is only minimally absorbed through the skin and is of low toxicity to humans, inhalation of sulfur dust or contact with eyes or skin may cause irritation. Excessive ingestion of sulfur can cause a burning sensation or diarrhea,[126] and cases of life-threatening metabolic acidosis have been reported after patients deliberately consumed sulfur as a folk remedy.[127][128]
Toxicity of sulfur compounds
When sulfur burns in air, it produces sulfur dioxide. In water, this gas produces sulfurous acid and sulfites; sulfites are antioxidants that inhibit growth of aerobic bacteria and a useful food additive in small amounts. At high concentrations these acids harm the lungs, eyes, or other tissues.[129] In organisms without lungs such as insects or plants, sulfite in high concentration prevents respiration.
Sulfur trioxide (made by catalysis from sulfur dioxide) and sulfuric acid are similarly highly acidic and corrosive in the presence of water. Concentrated sulfuric acid is a strong dehydrating agent that can strip available water molecules and water components from sugar and organic tissue.[130]
The burning of coal and/or petroleum by industry and power plants generates sulfur dioxide (SO2) that reacts with atmospheric water and oxygen to produce sulfuric acid (H2SO4) and sulfurous acid (H2SO3).[131] These acids are components of acid rain, lowering the pH of soil and freshwater bodies, sometimes resulting in substantial damage to the environment and chemical weathering of statues and structures. Fuel standards increasingly require that fuel producers extract sulfur from fossil fuels to prevent acid rain formation. This extracted and refined sulfur represents a large portion of sulfur production. In coal-fired power plants, flue gases are sometimes purified. More modern power plants that use synthesis gas extract the sulfur before they burn the gas.
Hydrogen sulfide is about one-half as toxic as hydrogen cyanide, and intoxicates by the same mechanism (inhibition of the respiratory enzyme cytochrome oxidase),[132] though hydrogen sulfide is less likely to cause sudden poisonings from small inhaled amounts (near its permissible exposure limit (PEL) of 20 ppm) because of its disagreeable odor.[133] However, its presence in ambient air at concentration over 100–150 ppm quickly deadens the sense of smell,[134] and a victim may breathe increasing quantities without noticing until severe symptoms cause death. Dissolved sulfide and hydrosulfide salts are toxic by the same mechanism.
See also
References
- ↑ "Standard Atomic Weights: Sulfur". CIAAW. 2009.
- ↑ Lide, D. R., ed. (2005). "Magnetic susceptibility of the elements and inorganic compounds". CRC Handbook of Chemistry and Physics (PDF) (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ↑ Weast, Robert (1984). CRC, Handbook of Chemistry and Physics. Boca Raton, Florida: Chemical Rubber Company Publishing. pp. E110. ISBN 0-8493-0464-4.
- ↑ "Sulfur History". Georgiagulfsulfur.com. Retrieved 12 February 2022.
- 1 2 3 Greenwood, N. N.; Earnshaw, A. (1997). Chemistry of the Elements (2nd ed.). Oxford: Butterworth-Heinemann. ISBN 0-7506-3365-4.
- ↑ Chisholm, Hugh, ed. (1911). . Encyclopædia Britannica. Vol. 4 (11th ed.). Cambridge University Press. p. 571.
- 1 2 3 4 Laurence Knight (19 July 2014). "Sulphur surplus: Up to our necks in a diabolical element". BBC.
- 1 2 "Sulfur". Elements. BBC. 11 October 2014.. Download here.
- ↑ Rettig, S. J.; Trotter, J. (15 December 1987). "Refinement of the structure of orthorhombic sulfur, α-S8" (PDF). Acta Crystallographica Section C. 43 (12): 2260–2262. doi:10.1107/S0108270187088152.
- ↑ A strong odor called "smell of sulfur" actually is given off by several sulfur compounds, such as hydrogen sulfide and organosulfur compounds.
- ↑ Tucker, Roy P. (1 January 1929). "Notes on the Sublimation of Sulfur between 25° and 50°C". Industrial & Engineering Chemistry. 21 (1): 44–47. doi:10.1021/ie50229a014. ISSN 0019-7866.
- 1 2 3 Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. pp. 645–665. ISBN 978-0-08-037941-8.
- ↑ Maldonado-Zagal, S. B.; Boden, P. J. (1 January 1982). "Hydrolysis of Elemental Sulphur in Water and its Effect on the Corrosion of Mild Steel". British Corrosion Journal. 17 (3): 116–120. doi:10.1179/000705982798274336. ISSN 0007-0599. Retrieved 23 June 2022.
- ↑ Munson, Ronald A. (February 1968). "The synthesis of iridium disulfide and nickel diarsenide having the pyrite structure" (PDF). Inorganic Chemistry. 7 (2): 389–390. doi:10.1021/ic50060a047.
- ↑ Egon Wiberg; Nils Wiberg (2001). Inorganic Chemistry. Academic Press. pp. 513–. ISBN 978-0-12-352651-9.
- ↑ Steudel, Ralf; Eckert, Bodo (2003). Solid Sulfur Allotropes Sulfur Allotropes. Topics in Current Chemistry. Vol. 230. pp. 1–80. doi:10.1007/b12110. ISBN 978-3-540-40191-9.
- ↑ Steudel, R. (1982). "Homocyclic sulfur molecules". Inorganic Ring Systems. Topics in Current Chemistry. Vol. 102. pp. 149–176. doi:10.1007/3-540-11345-2_10. ISBN 978-3-540-11345-4.
- ↑ Tebbe, Fred N.; Wasserman, E.; Peet, William G.; Vatvars, Arturs; Hayman, Alan C. (1982). "Composition of Elemental Sulfur in Solution: Equilibrium of S
6, S7, and S8 at Ambient Temperatures". Journal of the American Chemical Society. 104 (18): 4971–4972. doi:10.1021/ja00382a050. - ↑ Meyer, Beat (1964). "Solid Allotropes of Sulfur". Chemical Reviews. 64 (4): 429–451. doi:10.1021/cr60230a004.
- ↑ Meyer, Beat (1976). "Elemental sulfur". Chemical Reviews. 76 (3): 367–388. doi:10.1021/cr60301a003.
- ↑ Sulfur. Commission on Isotopic Abundances and Atomic Weights
- ↑ Haynes, William M., ed. (2011). CRC Handbook of Chemistry and Physics (92nd ed.). Boca Raton, FL: CRC Press. p. 1.14. ISBN 1-4398-5511-0.
- ↑ "Searching for the Origins of Presolar Grains". Energy.gov. Retrieved 4 February 2023.
- ↑ Paytan, Adina; Yao, Weiqi; Faul, Kristina; Gray, E.T. (2020). "Sulfur Isotope Stratigraphy". Geologic Time Scale. pp. 259–278. doi:10.1016/B978-0-12-824360-2.00009-7. ISBN 9780128243602.
- ↑ "NASA Astrobiology". astrobiology.nasa.gov. Retrieved 4 February 2023.
- ↑ Goldhaber, M.B.; Kaplan, I.R. (April 1980). "Mechanisms of sulfur incorporation and isotope fractionation during early diagenesis in sediments of the gulf of California". Marine Chemistry. 9 (2): 95–143. doi:10.1016/0304-4203(80)90063-8.
- ↑ Sim, Min Sub; Bosak, Tanja; Ono, Shuhei (July 2011). "Large Sulfur Isotope Fractionation Does Not Require Disproportionation". Science. 333 (6038): 74–77. doi:10.1126/science.1205103. ISSN 0036-8075. PMID 21719675. S2CID 1248182.
- ↑ Tsang, Man-Yin; Böttcher, Michael Ernst; Wortmann, Ulrich Georg (August 2023). "Estimating the effect of elemental sulfur disproportionation on the sulfur-isotope signatures in sediments". Chemical Geology. 632: 121533. doi:10.1016/j.chemgeo.2023.121533. S2CID 258600480.
- ↑ Kim, Ik Soo; Kwak, Seung Im; Park, Ul Jae; Bang, Hong Sik; Han, Hyun Soo (1 July 2005). Production of Sulfur-35 by the Cation Exchange Process. 2005 autumn meeting of the KNS, Busan (Korea, Republic of), 27–28 Oct 2005.
- ↑ "Sulfur-35 (35 S) safety information and specific handling precautions" (PDF). Yale Environmental Health & Safety.
- ↑ Cameron, A. G. W. (1957). "Stellar Evolution, Nuclear Astrophysics, and Nucleogenesis" (PDF). CRL-41.
- ↑ Mason, B. (1962). Meteorites. New York: John Wiley & Sons. p. 160. ISBN 978-0-908678-84-6.
- ↑ Lopes, Rosaly M. C.; Williams, David A. (2005). "Io after Galileo". Reports on Progress in Physics. 68 (2): 303–340. Bibcode:2005RPPh...68..303L. doi:10.1088/0034-4885/68/2/R02. S2CID 44208045.
- ↑ Rickwood, P. C. (1981). "The largest crystals" (PDF). American Mineralogist. 66: 885–907.
- ↑ Kutney, Gerald (2007). Sulfur: history, technology, applications & industry. Toronto: ChemTec. p. 43. ISBN 978-1-895198-37-9. OCLC 79256100.
- ↑ de Ronde, C. E. J.; Chadwick, W. W. Jr.; Ditchburn, R. G.; Embley, R. W.; Tunnicliffe, V.; Baker, E. T.; Walker, S. L.; Ferrini, V. L.; Merle, S. M. (2015). "Molten Sulfur Lakes of Intraoceanic Arc Volcanoes". Volcanic Lakes. Springer. pp. 261–288. doi:10.1007/978-3-642-36833-2. ISBN 978-3-642-36832-5.
- ↑ Klein, Cornelis; Hurlbut, Cornelius S. Jr. (1985). Manual of Mineralogy (20th ed.). Wiley. pp. 265–66. ISBN 0-471-80580-7.
- ↑ "Sulphur: Mineral information, data and localities". www.mindat.org.
- 1 2 3 4 5 Nehb, Wolfgang; Vydra, Karel (2006). "Sulfur". Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag. doi:10.1002/14356007.a25_507.pub2. ISBN 978-3-527-30673-2.
- ↑ Shriver, Atkins. Inorganic Chemistry, Fifth Edition. W. H. Freeman and Company, New York, 2010; pp 416
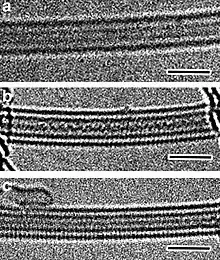
- ↑ Fujimori, Toshihiko; Morelos-Gómez, Aarón; Zhu, Zhen; Muramatsu, Hiroyuki; Futamura, Ryusuke; Urita, Koki; Terrones, Mauricio; Hayashi, Takuya; Endo, Morinobu; Young Hong, Sang; Chul Choi, Young; Tománek, David; Kaneko, Katsumi (2013). "Conducting linear chains of sulphur inside carbon nanotubes". Nature Communications. 4: 2162. Bibcode:2013NatCo...4.2162F. doi:10.1038/ncomms3162. PMC 3717502. PMID 23851903.
- ↑ Brauer, G., ed. (1963). Handbook of Preparative Inorganic Chemistry. Vol. 1 (2nd ed.). New York: Academic Press. p. 421.
- ↑ Hasek, W. R. (1961). "1,1,1-Trifluoroheptane". Organic Syntheses. 41: 104. doi:10.1002/0471264180.os041.28.
- 1 2 Lauss, H.-D.; Steffens, W. "Sulfur Halides". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a25_623. ISBN 978-3527306732.
- ↑ Vaughan, D. J.; Craig, J. R. (1978). Mineral Chemistry of Metal Sulfides. Cambridge: Cambridge University Press. ISBN 0-521-21489-0.
- ↑ Tsang, Man-Yin; Inagaki, Fumio (29 May 2020). "Microbial Life Deep Under the Seafloor—A Story of Not Giving Up". Frontiers for Young Minds. 8: 70. doi:10.3389/frym.2020.00070. ISSN 2296-6846.
- 1 2 Cremlyn, R. J. (1996). An Introduction to Organosulfur Chemistry. Chichester: John Wiley and Sons. ISBN 0-471-95512-4.
- ↑ Wilson, R. W.; Penzias, A. A.; Wannier, P. G.; Linke, R. A. (15 March 1976). "Isotopic abundances in interstellar carbon monosulfide". Astrophysical Journal. 204: L135–L137. Bibcode:1976ApJ...204L.135W. doi:10.1086/182072.
- ↑ Banoub, Joseph (2011). Detection of Biological Agents for the Prevention of Bioterrorism. NATO Science for Peace and Security Series A: Chemistry and Biology. p. 183. Bibcode:2011dbap.book.....B. doi:10.1007/978-90-481-9815-3. ISBN 978-90-481-9815-3. OCLC 697506461.
- ↑ "Sulfur in the Bible (14 instances)". bible.knowing-jesus.com. Retrieved 19 May 2022.
- ↑ Rapp, George Robert (4 February 2009). Archaeomineralogy. Springer. p. 242. ISBN 978-3-540-78593-4.
- ↑ Odyssey, book 22, lines 480–495. www.perseus.tufts.edu. Retrieved on 16 August 2012.
- ↑ Pliny the Elder on science and technology, John F. Healy, Oxford University Press, 1999, ISBN 0-19-814687-6, pp. 247–249.
- 1 2 3 Zhang, Yunming (1986). "The History of Science Society: Ancient Chinese Sulfur Manufacturing Processes". Isis. 77 (3): 487. doi:10.1086/354207. S2CID 144187385.
- ↑ Needham, Joseph; Yates, Robin (1994). Science and Civilisation in China, Volume 5: Chemistry and Chemical Technology, Part 6, Military Technology: Missiles and Sieges. Cambridge: Cambridge University Press. p. 120. ISBN 9780521327275. OCLC 489677531.
- ↑ Koch, Rudolf (1955). The book of signs : which contains all manner of symbols used from the earliest times to the Middle Ages by primitive peoples and early Christians. New York: Dover Publications. ISBN 0-486-20162-7.
- ↑ White, David Gordon (1996). The Alchemical Body — Siddha Traditions in Medieval India. Chicago: University of Chicago Press. pp. passim. ISBN 978-0-226-89499-7.
- ↑ Lin, A. N.; Reimer, R. J.; Carter, D. M. (1988). "Sulfur revisited". Journal of the American Academy of Dermatology. 18 (3): 553–558. doi:10.1016/S0190-9622(88)70079-1. PMID 2450900.
- ↑ Maibach, H. I.; Surber, C.; Orkin, M. (1990). "Sulfur revisited". Journal of the American Academy of Dermatology. 23 (1): 154–156. doi:10.1016/S0190-9622(08)81225-X. PMID 2365870.
- ↑ Gupta, A. K.; Nicol, K. (2004). "The use of sulfur in dermatology". Journal of Drugs in Dermatology. 3 (4): 427–31. PMID 15303787.
- ↑ Gupta, Aditya K; Nicol, Karyn (July–August 2004). "The Use of Sulfur in Dermatology". Journal of Drugs in Dermatology. 3 (4): 427–431. PMID 15303787.
- ↑ Donovan, Arthur (1996). Antoine Lavoisier: Science, Administration and Revolution. Cambridge University Press. p. 66. ISBN 978-0-521-56672-8.
- ↑ Poirier, Jean-Pierre (1998). Lavoisier: Chemist, Biologist, Economist. University of Pennsylvania Press. pp. 107–8. ISBN 978-0-8122-1649-3.
- ↑ Riall, Lucy (1998). Sicily and the Unification of Italy: Liberal Policy and Local Power, 1859–1866. Oxford University Press. ISBN 9780191542619. Retrieved 7 February 2013.
- ↑ Thomson, D. W. (April 1995). "Prelude to the Sulphur War of 1840: The Neapolitan Perspective". European History Quarterly. 25 (2): 163–180. doi:10.1177/026569149502500201. S2CID 145807900.
- ↑ Botsch, Walter (2001). "Chemiker, Techniker, Unternehmer: Zum 150. Geburtstag von Hermann Frasch". Chemie in unserer Zeit (in German). 35 (5): 324–331. doi:10.1002/1521-3781(200110)35:5<324::AID-CIUZ324>3.0.CO;2-9.
- ↑ Mass, Jennifer L; Anderson, Mark J (2003). "Pennsylvania German sulfur-inlaid furniture: characterization, reproduction, and ageing phenomena of the inlays". Measurement Science and Technology. 14 (9): 1598. doi:10.1088/0957-0233/14/9/311. ISSN 0957-0233. S2CID 250882259.
- ↑ Kogel, Jessica (2006). Industrial minerals & rocks: commodities, markets, and uses (7th ed.). Colorado: Littleton. p. 935. ISBN 978-0-87335-233-8. OCLC 62805047.
- ↑ "sulphur". Oxford English Dictionary (Online ed.). Oxford University Press. (Subscription or participating institution membership required.)
- ↑ "So long sulphur". Nature Chemistry. 1 (5): 333. 4 August 2009. Bibcode:2009NatCh...1Q.333.. doi:10.1038/nchem.301. PMID 21378874.
- ↑ McNaught, Alan (1991). "Journal style update". The Analyst. 116 (11): 1094. Bibcode:1991Ana...116.1094M. doi:10.1039/AN9911601094.
- ↑ "sulphur – definition of sulphur in English". Oxford Dictionaries. Archived from the original on 20 November 2016. Retrieved 19 November 2016.
- ↑ Riegel, Emil; Kent, James (2007). Kent and Riegel's Handbook of Industrial Chemistry and Biotechnology. Vol. 1. Springer. p. 1171. Bibcode:2007karh.book....... ISBN 978-0-387-27842-1. OCLC 74650396.
- ↑ Washington, Booker T. (1912). The Man Farthest Down: A Record of Observation and Study in Europe. Doubleday, Page. p. 214.
- ↑ McElvaney, Kevin (25 February 2015). "The Men Who Mine Volcanos". The Atlantic. Retrieved 26 February 2015.
- 1 2 3 Eow, John S. (2002). "Recovery of sulfur from sour acid gas: A review of the technology". Environmental Progress. 21 (3): 143–162. doi:10.1002/ep.670210312.
- 1 2 3 Schreiner, Bernhard (2008). "Der Claus-Prozess. Reich an Jahren und bedeutender denn je". Chemie in unserer Zeit. 42 (6): 378–392. doi:10.1002/ciuz.200800461.
- ↑ Hyndman, A. W.; Liu, J. K.; Denney, D. W. (1982). "Sulfur Recovery from Oil Sands". Sulfur: New Sources and Uses. ACS Symposium Series. Vol. 183. pp. 69–82. doi:10.1021/bk-1982-0183.ch005. ISBN 978-0-8412-0713-4.
- 1 2 Mohamed, Abdel-Mohsen Onsy; El-Gamal, Maisa M. (2010). Sulfur concrete for the construction industry: a sustainable development approach. Fort Lauderdale: J. Ross. pp. 104–105, 109. ISBN 978-1-60427-005-1. OCLC 531718953.
- ↑ Apodaca, Lori E. (2012) "Sulfur". Mineral Commodity Summaries. United States Geological Survey.
- 1 2 Apodaca, Lori E. "Mineral Yearbook 2010: Sulfur" (PDF). United States Geological Survey.
- 1 2 "FAQ – The Sulphur Institute". sulphurinstitute.org. The Sulphur Institute. 2020. Retrieved 27 February 2020.
- ↑ Zhao, F.; Hawkesford, M. J.; McGrath, S. P. (1999). "Sulphur Assimilation and Effects on Yield and Quality of Wheat". Journal of Cereal Science. 30 (1): 1–17. doi:10.1006/jcrs.1998.0241.
- ↑ Blake-Kalff, M. M. A. (2000). "Diagnosing sulfur deficiency in field-grown oilseed rape (Brassica napus L.) and wheat (Triticum aestivum L.)". Plant and Soil. 225 (1/2): 95–107. doi:10.1023/A:1026503812267. S2CID 44208638.
- ↑ Ceccotti, S. P. (1996). "Plant nutrient sulphur-a review of nutrient balance, environmental impact and fertilizers". Fertilizer Research. 43 (1–3): 117–125. doi:10.1007/BF00747690. S2CID 42207099.
- ↑ Glossary, United States: NASA Earth Observatory, acid rain, archived from the original on 13 December 2011, retrieved 15 February 2013
- ↑ Every, Richard L.; et al. (20 August 1968). "Method for Preparation of Wettable Sulfur" (PDF). Retrieved 20 May 2010.
- ↑ Hagers Handbuch der Pharmazeutischen Praxis (in German). Vol. 6B (4th ed.). Berlin–Heidelberg–New York: Springer. 1978. pp. 672–9. ISBN 978-3-540-07738-1.
- ↑ Arzneibuch-Kommentar. Wissenschaftliche Erläuterungen zum Europäischen Arzneibuch und zum Deutschen Arzneibuch [Pharmacopoeia Commentary. Scientific annotations to the European Pharmacopoeia and the German Pharmacopoeia] (in German) (23rd ed.). Stuttgart: Wissenschaftliche Verlagsgesellschaft. 2004. Monographie Schwefel zum äußerlichen Gebrauch [Monograph Sulfur for external use]. ISBN 978-3-8047-2575-1.
- ↑ Scott, Kevin A.; Njardarson, Jon T. (2019), Jiang, Xuefeng (ed.), "Analysis of US FDA-Approved Drugs Containing Sulfur Atoms" (PDF), Sulfur Chemistry, Topics in Current Chemistry Collections, Springer International Publishing, pp. 1–34, doi:10.1007/978-3-030-25598-5_1, ISBN 978-3-030-25598-5, retrieved 8 March 2023
- ↑ Pai, Rahul (10 February 2022). "Stabilization of gamma sulfur at room temperature to enable the use of carbonate electrolyte in Li–S batteries". Communications Chemistry. 5 (1): 17. doi:10.1038/s42004-022-00626-2. PMC 9814344. PMID 36697747. S2CID 246704531.
- ↑ "Sulphur and the Human Body". The Sulfur Institute. Retrieved 3 April 2021.
- ↑ "What is the body made of?". New Scientist. Archived from the original on 3 November 2021. Retrieved 9 November 2021.
- ↑ Helmenstine, Anne (3 February 2019). "Elemental Composition of the Human Body by Mass". ThoughtCo. Archived from the original on 13 April 2021. Retrieved 21 November 2021.
- ↑ Parcell, Stephen (February 2002). "Sulfur in human nutrition and applications in medicine". Alternative Medicine Review. 7 (1): 22–44. ISSN 1089-5159. PMID 11896744.
- ↑ Ingenbleek, Yves; Kimura, Hideo (July 2013). "Nutritional essentiality of sulfur in health and disease". Nutrition Reviews. 71 (7): 413–432. doi:10.1111/nure.12050. ISSN 1753-4887. PMID 23815141.
- ↑ Dworkin, Martin (March 2012). "Sergei Winogradsky: a founder of modern microbiology and the first microbial ecologist". FEMS Microbiology Reviews. 36 (2): 364–379. doi:10.1111/j.1574-6976.2011.00299.x. ISSN 1574-6976. PMID 22092289.
- ↑ Waksman, S. A.; Starkey, R. L. (20 January 1923). "On the Growth and Respiration of Sulfur-Oxidizing Bacteria". The Journal of General Physiology. 5 (3): 285–310. doi:10.1085/jgp.5.3.285. ISSN 0022-1295. PMC 2140527. PMID 19871997.
- ↑ Pronk JT; Meulenberg R; Hazeu W; Bos P; Kuenen JG (1990). "Oxidation of reduced inorganic sulphur compounds by acidophilic thiobacilli". FEMS Microbiology Letters. 75 (2–3): 293–306. doi:10.1111/j.1574-6968.1990.tb04103.x.
- ↑ Frigaard, Niels-Ulrik; Dahl, Christiane (1 January 2008), Poole, Robert K. (ed.), Sulfur Metabolism in Phototrophic Sulfur Bacteria, Advances in Microbial Physiology, vol. 54, Academic Press, pp. 103–200, doi:10.1016/S0065-2911(08)00002-7, ISBN 9780123743237, PMID 18929068, retrieved 17 May 2022
- ↑ Cavanaugh, Colleen M. (1994). "Microbial Symbiosis: Patterns of Diversity in the Marine Environment". American Zoologist. 34: 79–89. doi:10.1093/icb/34.1.79.
- ↑ Jones, Galen E.; Starkey, Robert L.; Feely, Herbert W.; Kulp, J. Laurence (22 June 1956). "Biological Origin of Native Sulfur in Salt Domes of Texas and Louisiana". Science. 123 (3208): 1124–1125. Bibcode:1956Sci...123.1124J. doi:10.1126/science.123.3208.1124. ISSN 0036-8075. PMID 17793426.
- ↑ Philip, G.; Wali, A. M. A.; Aref, M. A. M. (1 September 1994). "On the origin of native sulfur deposits in Gebel El Zeit, Gulf of Suez, Egypt". Carbonates and Evaporites. 9 (2): 223–232. doi:10.1007/BF03175232. ISSN 1878-5212. S2CID 128827551.
- ↑ "Petrography and mineralogy of the crystalline limestone of Fatha Formation from Mishraq area, Iraq". ResearchGate. Retrieved 15 April 2022.
- ↑ Heldt, Hans-Walter (1996). Pflanzenbiochemie (in German). Heidelberg: Spektrum Akademischer Verlag. pp. 321–333. ISBN 978-3-8274-0103-8.
- ↑ Kuenen, J. G.; Beudeker, R. F. (13 September 1982). "Microbiology of thiobacilli and other sulphur-oxidizing autotrophs, mixotrophs and heterotrophs". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 298 (1093): 473–497. Bibcode:1982RSPTB.298..473K. doi:10.1098/rstb.1982.0093. ISSN 0962-8436. PMID 6127737.
- ↑ Wasmund, Kenneth; Mußmann, Marc; Loy, Alexander (August 2017). "The life sulfuric: microbial ecology of sulfur cycling in marine sediments: Microbial sulfur cycling in marine sediments". Environmental Microbiology Reports. 9 (4): 323–344. doi:10.1111/1758-2229.12538. PMC 5573963. PMID 28419734.
- ↑ Gutiérrez-Preciado, A.; Romero, H.; Peimbert, M. (2010). "An Evolutionary Perspective on Amino Acids". Nature Education. 3 (9): 29.
- ↑ Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2002). Molecular Biology of the Cell. 4th edition. New York: Garland Science. ISBN 978-0-8153-3218-3.
- ↑ Arnér, Elias S. J.; Holmgren, Arne (25 December 2001). "Physiological functions of thioredoxin and thioredoxin reductase: Thioredoxin and thioredoxin reductase". European Journal of Biochemistry. 267 (20): 6102–6109. doi:10.1046/j.1432-1327.2000.01701.x. PMID 11012661.
- ↑ "Lysozyme". Drugs.com. Retrieved 19 May 2022.
- ↑ Nelson, D. L.; Cox, M. M. (2000). Lehninger, Principles of Biochemistry (3rd ed.). New York: Worth Publishing. ISBN 978-1-57259-153-0.
- ↑ Selhub, J. (1 July 1999). "Homocysteine metabolism". Annual Review of Nutrition. 19 (1): 217–246. doi:10.1146/annurev.nutr.19.1.217. ISSN 0199-9885. PMID 10448523.
- ↑ Huxtable, R. J. (1 January 1992). "Physiological actions of taurine". Physiological Reviews. 72 (1): 101–163. doi:10.1152/physrev.1992.72.1.101. ISSN 0031-9333. PMID 1731369.
- ↑ "The Function of Biotin". www.chem.uwec.edu. Retrieved 10 June 2022.
- ↑ Edwards, Katie A. "Thiamine Biochemistry". thiamine.dnr.cornell.edu. Retrieved 10 June 2022.
- ↑ Thauer, R. K. (1998). "Biochemistry of methanogenesis: a tribute to Marjory Stephenson:1998 Marjory Stephenson Prize Lecture". Microbiology. 144 (9): 2377–2406. doi:10.1099/00221287-144-9-2377. PMID 9782487.
- ↑ Pace, Nicholas J.; Weerapana, Eranthie (17 April 2014). "Zinc-binding cysteines: diverse functions and structural motifs". Biomolecules. 4 (2): 419–434. doi:10.3390/biom4020419. ISSN 2218-273X. PMC 4101490. PMID 24970223.
- ↑ Giles, Niroshini M; Watts, Aaron B; Giles, Gregory I; Fry, Fiona H; Littlechild, Jennifer A; Jacob, Claus (1 August 2003). "Metal and Redox Modulation of Cysteine Protein Function". Chemistry & Biology. 10 (8): 677–693. doi:10.1016/S1074-5521(03)00174-1. ISSN 1074-5521. PMID 12954327.
- ↑ Lippard, S. J.; Berg, J. M. (1994). Principles of Bioinorganic Chemistry. University Science Books. ISBN 978-0-935702-73-6.
- ↑ Mladenović, Dušan; Radosavljević, Tatjana; Hrnčić, Dragan; Rasic-Markovic, Aleksandra; Stanojlović, Olivera (26 July 2019). "The effects of dietary methionine restriction on the function and metabolic reprogramming in the liver and brain – implications for longevity". Reviews in the Neurosciences. 30 (6): 581–593. doi:10.1515/revneuro-2018-0073. ISSN 2191-0200. PMID 30817309. S2CID 73470156.
- ↑ Binz, Regina L.; Sadhukhan, Ratan; Miousse, Isabelle R.; Garg, Sarita; Koturbash, Igor; Zhou, Daohong; Hauer-Jensen, Martin; Pathak, Rupak (27 February 2021). "Dietary Methionine Deficiency Enhances Genetic Instability in Murine Immune Cells". International Journal of Molecular Sciences. 22 (5): 2378. doi:10.3390/ijms22052378. ISSN 1422-0067. PMC 7956689. PMID 33673497.
- ↑ Karakas, Erkan; Kisker, Caroline (18 October 2005). "Structural analysis of missense mutations causing isolated sulfite oxidase deficiency". Dalton Transactions (21): 3459–3463. doi:10.1039/B505789M. ISSN 1477-9234. PMID 16234925.
- ↑ "Sulfur 84683". S.
- ↑ "Chemical Datasheet. SULFUR, MOLTEN".
- ↑ "Sulfur General Fact Sheet". npic.orst.edu. Retrieved 2 September 2022.
- ↑ Schwartz, Steven M.; Carroll, Hugh M.; Scharschmidt, Linda A. (1 July 1986). "Sublimed (Inorganic) Sulfur Ingestion: A Cause of Life-Threatening Metabolic Acidosis With a High Anion Gap". Archives of Internal Medicine. 146 (7): 1437–1438. doi:10.1001/archinte.1986.00360190229034. ISSN 0003-9926. PMID 3718141.
- ↑ Blum, J. Eric; Coe, Fredric L. (13 January 2010). "Metabolic Acidosis after Sulfur Ingestion". New England Journal of Medicine. 297 (16): 869–870. doi:10.1056/nejm197710202971606. PMID 904661. Retrieved 2 September 2022.
- ↑ "Sulfur Dioxide | Toxicological Profile | ATSDR". Centers for Disease Control and Prevention. 26 March 2014. Retrieved 24 October 2023.
- ↑ Baker, Colin (1 March 2007). "The dehydration of sucrose". Education in Chemistry. Royal Society of Chemistry. Retrieved 14 June 2018.
- ↑ "4.1: Fossil Fuels and Products of Combustion". Engineering LibreTexts. 28 February 2021. Retrieved 24 October 2023.
- ↑ "Hydrogen Sulfide Toxicity: Practice Essentials, Pathophysiology, Etiology". Medscape. 30 March 2017 – via eMedicine.
- ↑ Summers, Vincent (8 April 2017). "Hydrogen Sulfide or Hydrogen Cyanide: Which is More Dangerous?". Quirky Science. Retrieved 23 August 2022.
- ↑ "Hydrogen Sulfide – Hazards | Occupational Safety and Health Administration". www.osha.gov. Retrieved 23 August 2022.
Further reading
Sigel, Astrid; Freisinger, Eva; Sigel, Roland K.O., eds. (2020). Transition Metals and Sulfur: A Strong Relationship for Life. Guest Editors Martha E Sosa Torres and Peter M.H.Kroneck. Berlin/Boston: de Gruyter. pp. xlv+455. ISBN 978-3-11-058889-7.
External links
- Sulfur at The Periodic Table of Videos (University of Nottingham)
- Atomic Data for Sulfur, NIST Physical Measurement Laboratory
- Sulfur phase diagram Archived 23 February 2010 at the Wayback Machine, Introduction to Chemistry for Ages 13–17
- Crystalline, liquid and polymerization of sulfur on Vulcano Island, Italy
- Sulfur and its use as a pesticide
- The Sulphur Institute
- Nutrient Stewardship and The Sulphur Institute