Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.
The first attempts to study whole transcriptomes began in the early 1990s. Subsequent technological advances since the late 1990s have repeatedly transformed the field and made transcriptomics a widespread discipline in biological sciences. There are two key contemporary techniques in the field: microarrays, which quantify a set of predetermined sequences, and RNA-Seq, which uses high-throughput sequencing to record all transcripts. As the technology improved, the volume of data produced by each transcriptome experiment increased. As a result, data analysis methods have steadily been adapted to more accurately and efficiently analyse increasingly large volumes of data. Transcriptome databases getting bigger and more useful as transcriptomes continue to be collected and shared by researchers. It would be almost impossible to interpret the information contained in a transcriptome without the knowledge of previous experiments.
Measuring the expression of an organism's genes in different tissues or conditions, or at different times, gives information on how genes are regulated and reveals details of an organism's biology. It can also be used to infer the functions of previously unannotated genes. Transcriptome analysis has enabled the study of how gene expression changes in different organisms and has been instrumental in the understanding of human disease. An analysis of gene expression in its entirety allows detection of broad coordinated trends which cannot be discerned by more targeted assays.
History
Transcriptomics has been characterised by the development of new techniques which have redefined what is possible every decade or so and rendered previous technologies obsolete. The first attempt at capturing a partial human transcriptome was published in 1991 and reported 609 mRNA sequences from the human brain.[2] In 2008, two human transcriptomes, composed of millions of transcript-derived sequences covering 16,000 genes, were published,[3][4] and by 2015 transcriptomes had been published for hundreds of individuals.[5][6] Transcriptomes of different disease states, tissues, or even single cells are now routinely generated.[6][7][8] This explosion in transcriptomics has been driven by the rapid development of new technologies with improved sensitivity and economy.[9][10][11][12]
Before transcriptomics
Studies of individual transcripts were being performed several decades before any transcriptomics approaches were available. Libraries of silkmoth mRNA transcripts were collected and converted to complementary DNA (cDNA) for storage using reverse transcriptase in the late 1970s.[13] In the 1980s, low-throughput sequencing using the Sanger method was used to sequence random transcripts, producing expressed sequence tags (ESTs).[2][14][15][16] The Sanger method of sequencing was predominant until the advent of high-throughput methods such as sequencing by synthesis (Solexa/Illumina). ESTs came to prominence during the 1990s as an efficient method to determine the gene content of an organism without sequencing the entire genome.[16] Amounts of individual transcripts were quantified using Northern blotting, nylon membrane arrays, and later reverse transcriptase quantitative PCR (RT-qPCR) methods,[17][18] but these methods are laborious and can only capture a tiny subsection of a transcriptome.[12] Consequently, the manner in which a transcriptome as a whole is expressed and regulated remained unknown until higher-throughput techniques were developed.
Early attempts
The word "transcriptome" was first used in the 1990s.[19][20] In 1995, one of the earliest sequencing-based transcriptomic methods was developed, serial analysis of gene expression (SAGE), which worked by Sanger sequencing of concatenated random transcript fragments.[21] Transcripts were quantified by matching the fragments to known genes. A variant of SAGE using high-throughput sequencing techniques, called digital gene expression analysis, was also briefly used.[9][22] However, these methods were largely overtaken by high throughput sequencing of entire transcripts, which provided additional information on transcript structure such as splice variants.[9]
Development of contemporary techniques
| RNA-Seq | Microarray | |
|---|---|---|
| Throughput | 1 day to 1 week per experiment[10] | 1–2 days per experiment[10] |
| Input RNA amount | Low ~ 1 ng total RNA[25] | High ~ 1 μg mRNA[26] |
| Labour intensity | High (sample preparation and data analysis)[10][23] | Low[10][23] |
| Prior knowledge | None required, although a reference genome/transcriptome sequence is useful[23] | Reference genome/transcriptome is required for design of probes[23] |
| Quantitation accuracy | ~90% (limited by sequence coverage)[27] | >90% (limited by fluorescence detection accuracy)[27] |
| Sequence resolution | RNA-Seq can detect SNPs and splice variants (limited by sequencing accuracy of ~99%)[27] | Specialised arrays can detect mRNA splice variants (limited by probe design and cross-hybridisation)[27] |
| Sensitivity | 1 transcript per million (approximate, limited by sequence coverage)[27] | 1 transcript per thousand (approximate, limited by fluorescence detection)[27] |
| Dynamic range | 100,000:1 (limited by sequence coverage)[28] | 1,000:1 (limited by fluorescence saturation)[28] |
| Technical reproducibility | >99%[29][30] | >99%[31][32] |
The dominant contemporary techniques, microarrays and RNA-Seq, were developed in the mid-1990s and 2000s.[9][33] Microarrays that measure the abundances of a defined set of transcripts via their hybridisation to an array of complementary probes were first published in 1995.[34][35] Microarray technology allowed the assay of thousands of transcripts simultaneously and at a greatly reduced cost per gene and labour saving.[36] Both spotted oligonucleotide arrays and Affymetrix high-density arrays were the method of choice for transcriptional profiling until the late 2000s.[12][33] Over this period, a range of microarrays were produced to cover known genes in model or economically important organisms. Advances in design and manufacture of arrays improved the specificity of probes and allowed more genes to be tested on a single array. Advances in fluorescence detection increased the sensitivity and measurement accuracy for low abundance transcripts.[35][37]
RNA-Seq is accomplished by reverse transcribing RNA in vitro and sequencing the resulting cDNAs.[10] Transcript abundance is derived from the number of counts from each transcript. The technique has therefore been heavily influenced by the development of high-throughput sequencing technologies.[9][11] Massively parallel signature sequencing (MPSS) was an early example based on generating 16–20 bp sequences via a complex series of hybridisations,[38][note 1] and was used in 2004 to validate the expression of ten thousand genes in Arabidopsis thaliana.[39] The earliest RNA-Seq work was published in 2006 with one hundred thousand transcripts sequenced using 454 technology.[40] This was sufficient coverage to quantify relative transcript abundance. RNA-Seq began to increase in popularity after 2008 when new Solexa/Illumina technologies allowed one billion transcript sequences to be recorded.[4][10][41][42] This yield now allows for the quantification and comparison of human transcriptomes.[43]
Data gathering
Generating data on RNA transcripts can be achieved via either of two main principles: sequencing of individual transcripts (ESTs, or RNA-Seq) or hybridisation of transcripts to an ordered array of nucleotide probes (microarrays).[23]
Isolation of RNA
All transcriptomic methods require RNA to first be isolated from the experimental organism before transcripts can be recorded. Although biological systems are incredibly diverse, RNA extraction techniques are broadly similar and involve mechanical disruption of cells or tissues, disruption of RNase with chaotropic salts,[44] disruption of macromolecules and nucleotide complexes, separation of RNA from undesired biomolecules including DNA, and concentration of the RNA via precipitation from solution or elution from a solid matrix.[44][45] Isolated RNA may additionally be treated with DNase to digest any traces of DNA.[46] It is necessary to enrich messenger RNA as total RNA extracts are typically 98% ribosomal RNA.[47] Enrichment for transcripts can be performed by poly-A affinity methods or by depletion of ribosomal RNA using sequence-specific probes.[48] Degraded RNA may affect downstream results; for example, mRNA enrichment from degraded samples will result in the depletion of 5’ mRNA ends and an uneven signal across the length of a transcript. Snap-freezing of tissue prior to RNA isolation is typical, and care is taken to reduce exposure to RNase enzymes once isolation is complete.[45]
Expressed sequence tags
An expressed sequence tag (EST) is a short nucleotide sequence generated from a single RNA transcript. RNA is first copied as complementary DNA (cDNA) by a reverse transcriptase enzyme before the resultant cDNA is sequenced.[16] Because ESTs can be collected without prior knowledge of the organism from which they come, they can be made from mixtures of organisms or environmental samples.[49][16] Although higher-throughput methods are now used, EST libraries commonly provided sequence information for early microarray designs; for example, a barley microarray was designed from 350,000 previously sequenced ESTs.[50]
Serial and cap analysis of gene expression (SAGE/CAGE)
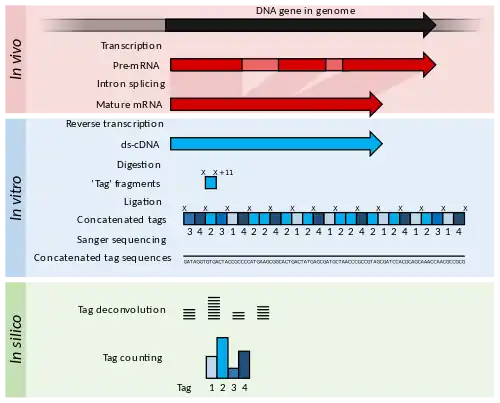
Serial analysis of gene expression (SAGE) was a development of EST methodology to increase the throughput of the tags generated and allow some quantitation of transcript abundance.[21] cDNA is generated from the RNA but is then digested into 11 bp "tag" fragments using restriction enzymes that cut DNA at a specific sequence, and 11 base pairs along from that sequence. These cDNA tags are then joined head-to-tail into long strands (>500 bp) and sequenced using low-throughput, but long read-length methods such as Sanger sequencing. The sequences are then divided back into their original 11 bp tags using computer software in a process called deconvolution.[21] If a high-quality reference genome is available, these tags may be matched to their corresponding gene in the genome. If a reference genome is unavailable, the tags can be directly used as diagnostic markers if found to be differentially expressed in a disease state.[21]
The cap analysis gene expression (CAGE) method is a variant of SAGE that sequences tags from the 5’ end of an mRNA transcript only.[52] Therefore, the transcriptional start site of genes can be identified when the tags are aligned to a reference genome. Identifying gene start sites is of use for promoter analysis and for the cloning of full-length cDNAs.
SAGE and CAGE methods produce information on more genes than was possible when sequencing single ESTs, but sample preparation and data analysis are typically more labour-intensive.[52]
Microarrays

Principles and advances
Microarrays usually consist of a grid of short nucleotide oligomers, known as "probes", typically arranged on a glass slide.[53] Transcript abundance is determined by hybridisation of fluorescently labelled transcripts to these probes.[54] The fluorescence intensity at each probe location on the array indicates the transcript abundance for that probe sequence.[54] Groups of probes designed to measure the same transcript (i.e., hybridizing a specific transcript in different positions) are usually referred to as "probesets".
Microarrays require some genomic knowledge from the organism of interest, for example, in the form of an annotated genome sequence, or a library of ESTs that can be used to generate the probes for the array.[36]
Methods
Microarrays for transcriptomics typically fall into one of two broad categories: low-density spotted arrays or high-density short probe arrays. Transcript abundance is inferred from the intensity of fluorescence derived from fluorophore-tagged transcripts that bind to the array.[36]
Spotted low-density arrays typically feature picolitre[note 2] drops of a range of purified cDNAs arrayed on the surface of a glass slide.[55] These probes are longer than those of high-density arrays and cannot identify alternative splicing events. Spotted arrays use two different fluorophores to label the test and control samples, and the ratio of fluorescence is used to calculate a relative measure of abundance.[56] High-density arrays use a single fluorescent label, and each sample is hybridised and detected individually.[57] High-density arrays were popularised by the Affymetrix GeneChip array, where each transcript is quantified by several short 25-mer probes that together assay one gene.[58]
NimbleGen arrays were a high-density array produced by a maskless-photochemistry method, which permitted flexible manufacture of arrays in small or large numbers. These arrays had 100,000s of 45 to 85-mer probes and were hybridised with a one-colour labelled sample for expression analysis.[59] Some designs incorporated up to 12 independent arrays per slide.
RNA-Seq
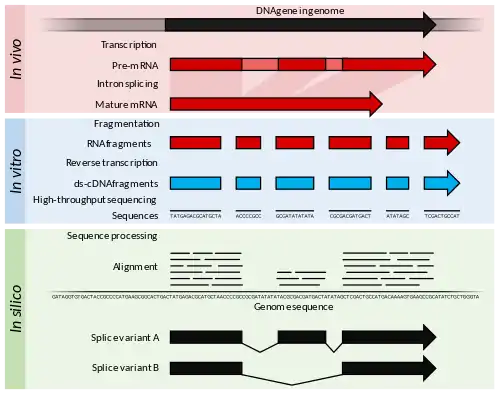
Principles and advances
RNA-Seq refers to the combination of a high-throughput sequencing methodology with computational methods to capture and quantify transcripts present in an RNA extract.[10] The nucleotide sequences generated are typically around 100 bp in length, but can range from 30 bp to over 10,000 bp depending on the sequencing method used. RNA-Seq leverages deep sampling of the transcriptome with many short fragments from a transcriptome to allow computational reconstruction of the original RNA transcript by aligning reads to a reference genome or to each other (de novo assembly).[9] Both low-abundance and high-abundance RNAs can be quantified in an RNA-Seq experiment (dynamic range of 5 orders of magnitude)—a key advantage over microarray transcriptomes. In addition, input RNA amounts are much lower for RNA-Seq (nanogram quantity) compared to microarrays (microgram quantity), which allow examination of the transcriptome even at a single-cell resolution when combined with amplification of cDNA.[25][60] Theoretically, there is no upper limit of quantification in RNA-Seq, and background noise is very low for 100 bp reads in non-repetitive regions.[10]
RNA-Seq may be used to identify genes within a genome, or identify which genes are active at a particular point in time, and read counts can be used to accurately model the relative gene expression level. RNA-Seq methodology has constantly improved, primarily through the development of DNA sequencing technologies to increase throughput, accuracy, and read length.[61] Since the first descriptions in 2006 and 2008,[40][62] RNA-Seq has been rapidly adopted and overtook microarrays as the dominant transcriptomics technique in 2015.[63]
The quest for transcriptome data at the level of individual cells has driven advances in RNA-Seq library preparation methods, resulting in dramatic advances in sensitivity. Single-cell transcriptomes are now well described and have even been extended to in situ RNA-Seq where transcriptomes of individual cells are directly interrogated in fixed tissues.[64]
Methods
RNA-Seq was established in concert with the rapid development of a range of high-throughput DNA sequencing technologies.[65] However, before the extracted RNA transcripts are sequenced, several key processing steps are performed. Methods differ in the use of transcript enrichment, fragmentation, amplification, single or paired-end sequencing, and whether to preserve strand information.[65]
The sensitivity of an RNA-Seq experiment can be increased by enriching classes of RNA that are of interest and depleting known abundant RNAs. The mRNA molecules can be separated using oligonucleotides probes which bind their poly-A tails. Alternatively, ribo-depletion can be used to specifically remove abundant but uninformative ribosomal RNAs (rRNAs) by hybridisation to probes tailored to the taxon's specific rRNA sequences (e.g. mammal rRNA, plant rRNA). However, ribo-depletion can also introduce some bias via non-specific depletion of off-target transcripts.[66] Small RNAs, such as micro RNAs, can be purified based on their size by gel electrophoresis and extraction.
Since mRNAs are longer than the read-lengths of typical high-throughput sequencing methods, transcripts are usually fragmented prior to sequencing.[67] The fragmentation method is a key aspect of sequencing library construction. Fragmentation may be achieved by chemical hydrolysis, nebulisation, sonication, or reverse transcription with chain-terminating nucleotides.[67] Alternatively, fragmentation and cDNA tagging may be done simultaneously by using transposase enzymes.[68]
During preparation for sequencing, cDNA copies of transcripts may be amplified by PCR to enrich for fragments that contain the expected 5’ and 3’ adapter sequences.[69] Amplification is also used to allow sequencing of very low input amounts of RNA, down to as little as 50 pg in extreme applications.[70] Spike-in controls of known RNAs can be used for quality control assessment to check library preparation and sequencing, in terms of GC-content, fragment length, as well as the bias due to fragment position within a transcript.[71] Unique molecular identifiers (UMIs) are short random sequences that are used to individually tag sequence fragments during library preparation so that every tagged fragment is unique.[72] UMIs provide an absolute scale for quantification, the opportunity to correct for subsequent amplification bias introduced during library construction, and accurately estimate the initial sample size. UMIs are particularly well-suited to single-cell RNA-Seq transcriptomics, where the amount of input RNA is restricted and extended amplification of the sample is required.[73][74][75]
Once the transcript molecules have been prepared they can be sequenced in just one direction (single-end) or both directions (paired-end). A single-end sequence is usually quicker to produce, cheaper than paired-end sequencing and sufficient for quantification of gene expression levels. Paired-end sequencing produces more robust alignments/assemblies, which is beneficial for gene annotation and transcript isoform discovery.[10] Strand-specific RNA-Seq methods preserve the strand information of a sequenced transcript.[76] Without strand information, reads can be aligned to a gene locus but do not inform in which direction the gene is transcribed. Stranded-RNA-Seq is useful for deciphering transcription for genes that overlap in different directions and to make more robust gene predictions in non-model organisms.[76]
| Platform | Commercial release | Typical read length | Maximum throughput per run | Single read accuracy | RNA-Seq runs deposited in the NCBI SRA (Oct 2016)[79] |
|---|---|---|---|---|---|
| 454 Life Sciences | 2005 | 700 bp | 0.7 Gbp | 99.9% | 3548 |
| Illumina | 2006 | 50–300 bp | 900 Gbp | 99.9% | 362903 |
| SOLiD | 2008 | 50 bp | 320 Gbp | 99.9% | 7032 |
| Ion Torrent | 2010 | 400 bp | 30 Gbp | 98% | 1953 |
| PacBio | 2011 | 10,000 bp | 2 Gbp | 87% | 160 |
Legend: NCBI SRA – National center for biotechnology information sequence read archive.
Currently RNA-Seq relies on copying RNA molecules into cDNA molecules prior to sequencing; therefore, the subsequent platforms are the same for transcriptomic and genomic data. Consequently, the development of DNA sequencing technologies has been a defining feature of RNA-Seq.[78][80][81] Direct sequencing of RNA using nanopore sequencing represents a current state-of-the-art RNA-Seq technique.[82][83] Nanopore sequencing of RNA can detect modified bases that would be otherwise masked when sequencing cDNA and also eliminates amplification steps that can otherwise introduce bias.[11][84]
The sensitivity and accuracy of an RNA-Seq experiment are dependent on the number of reads obtained from each sample.[85][86] A large number of reads are needed to ensure sufficient coverage of the transcriptome, enabling detection of low abundance transcripts. Experimental design is further complicated by sequencing technologies with a limited output range, the variable efficiency of sequence creation, and variable sequence quality. Added to those considerations is that every species has a different number of genes and therefore requires a tailored sequence yield for an effective transcriptome. Early studies determined suitable thresholds empirically, but as the technology matured suitable coverage was predicted computationally by transcriptome saturation. Somewhat counter-intuitively, the most effective way to improve detection of differential expression in low expression genes is to add more biological replicates rather than adding more reads.[87] The current benchmarks recommended by the Encyclopedia of DNA Elements (ENCODE) Project are for 70-fold exome coverage for standard RNA-Seq and up to 500-fold exome coverage to detect rare transcripts and isoforms.[88][89][90]
Data analysis
Transcriptomics methods are highly parallel and require significant computation to produce meaningful data for both microarray and RNA-Seq experiments.[91][92][93][94][95] Microarray data is recorded as high-resolution images, requiring feature detection and spectral analysis.[96] Microarray raw image files are each about 750 MB in size, while the processed intensities are around 60 MB in size. Multiple short probes matching a single transcript can reveal details about the intron-exon structure, requiring statistical models to determine the authenticity of the resulting signal. RNA-Seq studies produce billions of short DNA sequences, which must be aligned to reference genomes composed of millions to billions of base pairs. De novo assembly of reads within a dataset requires the construction of highly complex sequence graphs.[97] RNA-Seq operations are highly repetitious and benefit from parallelised computation but modern algorithms mean consumer computing hardware is sufficient for simple transcriptomics experiments that do not require de novo assembly of reads.[98] A human transcriptome could be accurately captured using RNA-Seq with 30 million 100 bp sequences per sample.[85][86] This example would require approximately 1.8 gigabytes of disk space per sample when stored in a compressed fastq format. Processed count data for each gene would be much smaller, equivalent to processed microarray intensities. Sequence data may be stored in public repositories, such as the Sequence Read Archive (SRA).[99] RNA-Seq datasets can be uploaded via the Gene Expression Omnibus.[100]
Image processing

Microarray image processing must correctly identify the regular grid of features within an image and independently quantify the fluorescence intensity for each feature. Image artefacts must be additionally identified and removed from the overall analysis. Fluorescence intensities directly indicate the abundance of each sequence, since the sequence of each probe on the array is already known.[102]
The first steps of RNA-seq also include similar image processing; however, conversion of images to sequence data is typically handled automatically by the instrument software. The Illumina sequencing-by-synthesis method results in an array of clusters distributed over the surface of a flow cell.[103] The flow cell is imaged up to four times during each sequencing cycle, with tens to hundreds of cycles in total. Flow cell clusters are analogous to microarray spots and must be correctly identified during the early stages of the sequencing process. In Roche’s pyrosequencing method, the intensity of emitted light determines the number of consecutive nucleotides in a homopolymer repeat. There are many variants on these methods, each with a different error profile for the resulting data.[104]
RNA-Seq data analysis
RNA-Seq experiments generate a large volume of raw sequence reads which have to be processed to yield useful information. Data analysis usually requires a combination of bioinformatics software tools (see also List of RNA-Seq bioinformatics tools) that vary according to the experimental design and goals. The process can be broken down into four stages: quality control, alignment, quantification, and differential expression.[105] Most popular RNA-Seq programs are run from a command-line interface, either in a Unix environment or within the R/Bioconductor statistical environment.[94]
Quality control
Sequence reads are not perfect, so the accuracy of each base in the sequence needs to be estimated for downstream analyses. Raw data is examined to ensure: quality scores for base calls are high, the GC content matches the expected distribution, short sequence motifs (k-mers) are not over-represented, and the read duplication rate is acceptably low.[86] Several software options exist for sequence quality analysis, including FastQC and FaQCs.[106][107] Abnormalities may be removed (trimming) or tagged for special treatment during later processes.
Alignment
In order to link sequence read abundance to the expression of a particular gene, transcript sequences are aligned to a reference genome or de novo aligned to one another if no reference is available.[108][109][110] The key challenges for alignment software include sufficient speed to permit billions of short sequences to be aligned in a meaningful timeframe, flexibility to recognise and deal with intron splicing of eukaryotic mRNA, and correct assignment of reads that map to multiple locations. Software advances have greatly addressed these issues, and increases in sequencing read length reduce the chance of ambiguous read alignments. A list of currently available high-throughput sequence aligners is maintained by the EBI.[111][112]
Alignment of primary transcript mRNA sequences derived from eukaryotes to a reference genome requires specialised handling of intron sequences, which are absent from mature mRNA.[113] Short read aligners perform an additional round of alignments specifically designed to identify splice junctions, informed by canonical splice site sequences and known intron splice site information. Identification of intron splice junctions prevents reads from being misaligned across splice junctions or erroneously discarded, allowing more reads to be aligned to the reference genome and improving the accuracy of gene expression estimates. Since gene regulation may occur at the mRNA isoform level, splice-aware alignments also permit detection of isoform abundance changes that would otherwise be lost in a bulked analysis.[114]
De novo assembly can be used to align reads to one another to construct full-length transcript sequences without use of a reference genome.[115] Challenges particular to de novo assembly include larger computational requirements compared to a reference-based transcriptome, additional validation of gene variants or fragments, and additional annotation of assembled transcripts. The first metrics used to describe transcriptome assemblies, such as N50, have been shown to be misleading[116] and improved evaluation methods are now available.[117][118] Annotation-based metrics are better assessments of assembly completeness, such as contig reciprocal best hit count. Once assembled de novo, the assembly can be used as a reference for subsequent sequence alignment methods and quantitative gene expression analysis.
| Software | Released | Last updated | Computational efficiency | Strengths and weaknesses |
|---|---|---|---|---|
| Velvet-Oases[119][120] | 2008 | 2011 | Low, single-threaded, high RAM requirement | The original short read assembler. It is now largely superseded. |
| SOAPdenovo-trans[109] | 2011 | 2014 | Moderate, multi-thread, medium RAM requirement | An early example of a short read assembler. It has been updated for transcriptome assembly. |
| Trans-ABySS[121] | 2010 | 2016 | Moderate, multi-thread, medium RAM requirement | Suited to short reads, can handle complex transcriptomes, and an MPI-parallel version is available for computing clusters. |
| Trinity[122][97] | 2011 | 2017 | Moderate, multi-thread, medium RAM requirement | Suited to short reads. It can handle complex transcriptomes but is memory intensive. |
| miraEST[123] | 1999 | 2016 | Moderate, multi-thread, medium RAM requirement | Can process repetitive sequences, combine different sequencing formats, and a wide range of sequence platforms are accepted. |
| Newbler[124] | 2004 | 2012 | Low, single-thread, high RAM requirement | Specialised to accommodate the homo-polymer sequencing errors typical of Roche 454 sequencers. |
| CLC genomics workbench[125] | 2008 | 2014 | High, multi-thread, low RAM requirement | Has a graphical user interface, can combine diverse sequencing technologies, has no transcriptome-specific features, and a licence must be purchased before use. |
| SPAdes[126] | 2012 | 2017 | High, multi-thread, low RAM requirement | Used for transcriptomics experiments on single cells. |
| RSEM[127] | 2011 | 2017 | High, multi-thread, low RAM requirement | Can estimate frequency of alternatively spliced transcripts. User friendly. |
| StringTie[98][128] | 2015 | 2019 | High, multi-thread, low RAM requirement | Can use a combination of reference-guided and de novo assembly methods to identify transcripts. |
Legend: RAM – random access memory; MPI – message passing interface; EST – expressed sequence tag.
Quantification
Quantification of sequence alignments may be performed at the gene, exon, or transcript level.[91][87] Typical outputs include a table of read counts for each feature supplied to the software; for example, for genes in a general feature format file. Gene and exon read counts may be calculated quite easily using HTSeq, for example.[130] Quantitation at the transcript level is more complicated and requires probabilistic methods to estimate transcript isoform abundance from short read information; for example, using cufflinks software.[114] Reads that align equally well to multiple locations must be identified and either removed, aligned to one of the possible locations, or aligned to the most probable location.
Some quantification methods can circumvent the need for an exact alignment of a read to a reference sequence altogether. The kallisto software method combines pseudoalignment and quantification into a single step that runs 2 orders of magnitude faster than contemporary methods such as those used by tophat/cufflinks software, with less computational burden.[131]
Differential expression
Once quantitative counts of each transcript are available, differential gene expression is measured by normalising, modelling, and statistically analysing the data.[108] Most tools will read a table of genes and read counts as their input, but some programs, such as cuffdiff, will accept binary alignment map format read alignments as input. The final outputs of these analyses are gene lists with associated pair-wise tests for differential expression between treatments and the probability estimates of those differences.[132]
| Software | Environment | Specialisation |
|---|---|---|
| Cuffdiff2[108] | Unix-based | Transcript analysis that tracks alternative splicing of mRNA |
| EdgeR[93] | R/Bioconductor | Any count-based genomic data |
| DEseq2[133] | R/Bioconductor | Flexible data types, low replication |
| Limma/Voom[92] | R/Bioconductor | Microarray or RNA-Seq data, flexible experiment design |
| Ballgown[134] | R/Bioconductor | Efficient and sensitive transcript discovery, flexible. |
Legend: mRNA - messenger RNA.
Validation
Transcriptomic analyses may be validated using an independent technique, for example, quantitative PCR (qPCR), which is recognisable and statistically assessable.[135] Gene expression is measured against defined standards both for the gene of interest and control genes. The measurement by qPCR is similar to that obtained by RNA-Seq wherein a value can be calculated for the concentration of a target region in a given sample. qPCR is, however, restricted to amplicons smaller than 300 bp, usually toward the 3’ end of the coding region, avoiding the 3’UTR.[136] If validation of transcript isoforms is required, an inspection of RNA-Seq read alignments should indicate where qPCR primers might be placed for maximum discrimination. The measurement of multiple control genes along with the genes of interest produces a stable reference within a biological context.[137] qPCR validation of RNA-Seq data has generally shown that different RNA-Seq methods are highly correlated.[62][138][139]
Functional validation of key genes is an important consideration for post transcriptome planning. Observed gene expression patterns may be functionally linked to a phenotype by an independent knock-down/rescue study in the organism of interest.[140]
Applications
Diagnostics and disease profiling
Transcriptomic strategies have seen broad application across diverse areas of biomedical research, including disease diagnosis and profiling.[10][141] RNA-Seq approaches have allowed for the large-scale identification of transcriptional start sites, uncovered alternative promoter usage, and novel splicing alterations. These regulatory elements are important in human disease and, therefore, defining such variants is crucial to the interpretation of disease-association studies.[142] RNA-Seq can also identify disease-associated single nucleotide polymorphisms (SNPs), allele-specific expression, and gene fusions, which contributes to the understanding of disease causal variants.[143]
Retrotransposons are transposable elements which proliferate within eukaryotic genomes through a process involving reverse transcription. RNA-Seq can provide information about the transcription of endogenous retrotransposons that may influence the transcription of neighboring genes by various epigenetic mechanisms that lead to disease.[144] Similarly, the potential for using RNA-Seq to understand immune-related disease is expanding rapidly due to the ability to dissect immune cell populations and to sequence T cell and B cell receptor repertoires from patients.[145][146]
Human and pathogen transcriptomes
RNA-Seq of human pathogens has become an established method for quantifying gene expression changes, identifying novel virulence factors, predicting antibiotic resistance, and unveiling host-pathogen immune interactions.[147][148] A primary aim of this technology is to develop optimised infection control measures and targeted individualised treatment.[146]
Transcriptomic analysis has predominantly focused on either the host or the pathogen. Dual RNA-Seq has been applied to simultaneously profile RNA expression in both the pathogen and host throughout the infection process. This technique enables the study of the dynamic response and interspecies gene regulatory networks in both interaction partners from initial contact through to invasion and the final persistence of the pathogen or clearance by the host immune system.[149][150]
Responses to environment
Transcriptomics allows identification of genes and pathways that respond to and counteract biotic and abiotic environmental stresses.[151][140] The non-targeted nature of transcriptomics allows the identification of novel transcriptional networks in complex systems. For example, comparative analysis of a range of chickpea lines at different developmental stages identified distinct transcriptional profiles associated with drought and salinity stresses, including identifying the role of transcript isoforms of AP2-EREBP.[151] Investigation of gene expression during biofilm formation by the fungal pathogen Candida albicans revealed a co-regulated set of genes critical for biofilm establishment and maintenance.[152]
Transcriptomic profiling also provides crucial information on mechanisms of drug resistance. Analysis of over 1000 isolates of Plasmodium falciparum, a virulent parasite responsible for malaria in humans,[153] identified that upregulation of the unfolded protein response and slower progression through the early stages of the asexual intraerythrocytic developmental cycle were associated with artemisinin resistance in isolates from Southeast Asia.[154]
The use of transcriptomics is also important to investigate responses in the marine environment.[155] In marine ecology, "stress" and "adaptation" have been among the most common research topics, especially related to anthropogenic stress, such as global change and pollution.[155] Most of the studies in this area have been done in animals, although invertebrates have been underrepresented.[155] One issue still is a deficiency in functional genetic studies, which hamper gene annotations, especially for non-model species, and can lead to vague conclusions on the effects of responses studied.[155]
Gene function annotation
All transcriptomic techniques have been particularly useful in identifying the functions of genes and identifying those responsible for particular phenotypes. Transcriptomics of Arabidopsis ecotypes that hyperaccumulate metals correlated genes involved in metal uptake, tolerance, and homeostasis with the phenotype.[156] Integration of RNA-Seq datasets across different tissues has been used to improve annotation of gene functions in commercially important organisms (e.g. cucumber)[157] or threatened species (e.g. koala).[158]
Assembly of RNA-Seq reads is not dependent on a reference genome[122] and so is ideal for gene expression studies of non-model organisms with non-existing or poorly developed genomic resources. For example, a database of SNPs used in Douglas fir breeding programs was created by de novo transcriptome analysis in the absence of a sequenced genome.[159] Similarly, genes that function in the development of cardiac, muscle, and nervous tissue in lobsters were identified by comparing the transcriptomes of the various tissue types without use of a genome sequence.[160] RNA-Seq can also be used to identify previously unknown protein coding regions in existing sequenced genomes.
A transcriptome based aging clock
Aging-related preventive interventions are not possible without personal aging speed measurement. The most up to date and complex way to measure aging rate is by using varying biomarkers of human aging is based on the utilization of deep neural networks which may be trained on any type of omics biological data to predict the subject's age. Aging has been shown to be a strong driver of transcriptome changes.[161][162] Aging clocks based on transcriptomes have suffered from considerable variation in the data and relatively low accuracy. However an approach that uses temporal scaling and binarization of transcriptomes to define a gene set that predicts biological age with an accuracy allowed to reach an assessment close to the theoretical limit.[161]
Non-coding RNA
Transcriptomics is most commonly applied to the mRNA content of the cell. However, the same techniques are equally applicable to non-coding RNAs (ncRNAs) that are not translated into a protein, but instead have direct functions (e.g. roles in protein translation, DNA replication, RNA splicing, and transcriptional regulation).[163][164][165][166] Many of these ncRNAs affect disease states, including cancer, cardiovascular, and neurological diseases.[167]
Transcriptome databases
Transcriptomics studies generate large amounts of data that have potential applications far beyond the original aims of an experiment. As such, raw or processed data may be deposited in public databases to ensure their utility for the broader scientific community. For example, as of 2018, the Gene Expression Omnibus contained millions of experiments.[168]
| Name | Host | Data | Description |
|---|---|---|---|
| Gene Expression Omnibus[100] | NCBI | Microarray RNA-Seq | First transcriptomics database to accept data from any source. Introduced MIAME and MINSEQE community standards that define necessary experiment metadata to ensure effective interpretation and repeatability.[169][170] |
| ArrayExpress[171] | ENA | Microarray | Imports datasets from the Gene Expression Omnibus and accepts direct submissions. Processed data and experiment metadata is stored at ArrayExpress, while the raw sequence reads are held at the ENA. Complies with MIAME and MINSEQE standards.[169][170] |
| Expression Atlas[172] | EBI | Microarray RNA-Seq | Tissue-specific gene expression database for animals and plants. Displays secondary analyses and visualisation, such as functional enrichment of Gene Ontology terms, InterPro domains, or pathways. Links to protein abundance data where available. |
| Genevestigator[173] | Privately curated | Microarray RNA-Seq | Contains manual curations of public transcriptome datasets, focusing on medical and plant biology data. Individual experiments are normalised across the full database to allow comparison of gene expression across diverse experiments. Full functionality requires licence purchase, with free access to a limited functionality. |
| RefEx[174] | DDBJ | All | Human, mouse, and rat transcriptomes from 40 different organs. Gene expression visualised as heatmaps projected onto 3D representations of anatomical structures. |
| NONCODE[175] | noncode.org | RNA-Seq | Non-coding RNAs (ncRNAs) excluding tRNA and rRNA. |
Legend: NCBI – National Center for Biotechnology Information; EBI – European Bioinformatics Institute; DDBJ – DNA Data Bank of Japan; ENA – European Nucleotide Archive; MIAME – Minimum Information About a Microarray Experiment; MINSEQE – Minimum Information about a high-throughput nucleotide SEQuencing Experiment.
See also
References
![]() This article was adapted from the following source under a CC BY 4.0 license (2017) (reviewer reports):
Rohan Lowe; Neil Shirley; Mark Bleackley; Stephen Dolan; Thomas Shafee (18 May 2017). "Transcriptomics technologies". PLOS Computational Biology. 13 (5): e1005457. doi:10.1371/JOURNAL.PCBI.1005457. ISSN 1553-734X. PMC 5436640. PMID 28545146. S2CID 3714586. Wikidata Q33703532.
This article was adapted from the following source under a CC BY 4.0 license (2017) (reviewer reports):
Rohan Lowe; Neil Shirley; Mark Bleackley; Stephen Dolan; Thomas Shafee (18 May 2017). "Transcriptomics technologies". PLOS Computational Biology. 13 (5): e1005457. doi:10.1371/JOURNAL.PCBI.1005457. ISSN 1553-734X. PMC 5436640. PMID 28545146. S2CID 3714586. Wikidata Q33703532.{{cite journal}}: CS1 maint: unflagged free DOI (link)
- ↑ "Medline trend: automated yearly statistics of PubMed results for any query". dan.corlan.net. Retrieved 2016-10-05.
- 1 2 Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, et al. (June 1991). "Complementary DNA sequencing: expressed sequence tags and human genome project". Science. 252 (5013): 1651–6. Bibcode:1991Sci...252.1651A. doi:10.1126/science.2047873. PMID 2047873. S2CID 13436211.
- ↑ Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (December 2008). "Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing". Nature Genetics. 40 (12): 1413–5. doi:10.1038/ng.259. PMID 18978789. S2CID 9228930.
- 1 2 Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, et al. (August 2008). "A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome". Science. 321 (5891): 956–60. Bibcode:2008Sci...321..956S. doi:10.1126/science.1160342. PMID 18599741. S2CID 10013179.
- ↑ Lappalainen T, Sammeth M, Friedländer MR, 't Hoen PA, Monlong J, Rivas MA, et al. (September 2013). "Transcriptome and genome sequencing uncovers functional variation in humans". Nature. 501 (7468): 506–11. Bibcode:2013Natur.501..506L. doi:10.1038/nature12531. PMC 3918453. PMID 24037378.
- 1 2 Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. (May 2015). "Human genomics. The human transcriptome across tissues and individuals". Science. 348 (6235): 660–5. Bibcode:2015Sci...348..660M. doi:10.1126/science.aaa0355. PMC 4547472. PMID 25954002.
- ↑ Sandberg R (January 2014). "Entering the era of single-cell transcriptomics in biology and medicine". Nature Methods. 11 (1): 22–4. doi:10.1038/nmeth.2764. PMID 24524133. S2CID 27632439.
- ↑ Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA (May 2015). "The technology and biology of single-cell RNA sequencing". Molecular Cell. 58 (4): 610–20. doi:10.1016/j.molcel.2015.04.005. PMID 26000846.
- 1 2 3 4 5 6 McGettigan PA (February 2013). "Transcriptomics in the RNA-seq era". Current Opinion in Chemical Biology. 17 (1): 4–11. doi:10.1016/j.cbpa.2012.12.008. PMID 23290152.
- 1 2 3 4 5 6 7 8 9 10 11 12 Wang Z, Gerstein M, Snyder M (January 2009). "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
- 1 2 3 Ozsolak F, Milos PM (February 2011). "RNA sequencing: advances, challenges and opportunities". Nature Reviews Genetics. 12 (2): 87–98. doi:10.1038/nrg2934. PMC 3031867. PMID 21191423.
- 1 2 3 Morozova O, Hirst M, Marra MA (2009). "Applications of new sequencing technologies for transcriptome analysis". Annual Review of Genomics and Human Genetics. 10: 135–51. doi:10.1146/annurev-genom-082908-145957. PMID 19715439.
- ↑ Sim GK, Kafatos FC, Jones CW, Koehler MD, Efstratiadis A, Maniatis T (December 1979). "Use of a cDNA library for studies on evolution and developmental expression of the chorion multigene families". Cell. 18 (4): 1303–16. doi:10.1016/0092-8674(79)90241-1. PMID 519770.
- ↑ Sutcliffe JG, Milner RJ, Bloom FE, Lerner RA (August 1982). "Common 82-nucleotide sequence unique to brain RNA". Proceedings of the National Academy of Sciences of the United States of America. 79 (16): 4942–6. Bibcode:1982PNAS...79.4942S. doi:10.1073/pnas.79.16.4942. PMC 346801. PMID 6956902.
- ↑ Putney SD, Herlihy WC, Schimmel P (April 1983). "A new troponin T and cDNA clones for 13 different muscle proteins, found by shotgun sequencing". Nature. 302 (5910): 718–21. Bibcode:1983Natur.302..718P. doi:10.1038/302718a0. PMID 6687628. S2CID 4364361.
- 1 2 3 4 Marra MA, Hillier L, Waterston RH (January 1998). "Expressed sequence tags—ESTablishing bridges between genomes". Trends in Genetics. 14 (1): 4–7. doi:10.1016/S0168-9525(97)01355-3. PMID 9448457.
- ↑ Alwine JC, Kemp DJ, Stark GR (December 1977). "Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes". Proceedings of the National Academy of Sciences of the United States of America. 74 (12): 5350–4. Bibcode:1977PNAS...74.5350A. doi:10.1073/pnas.74.12.5350. PMC 431715. PMID 414220.
- ↑ Becker-André M, Hahlbrock K (November 1989). "Absolute mRNA quantification using the polymerase chain reaction (PCR). A novel approach by a PCR aided transcript titration assay (PATTY)". Nucleic Acids Research. 17 (22): 9437–46. doi:10.1093/nar/17.22.9437. PMC 335144. PMID 2479917.
- ↑ Piétu G, Mariage-Samson R, Fayein NA, Matingou C, Eveno E, Houlgatte R, Decraene C, Vandenbrouck Y, Tahi F, Devignes MD, Wirkner U, Ansorge W, Cox D, Nagase T, Nomura N, Auffray C (February 1999). "The Genexpress IMAGE knowledge base of the human brain transcriptome: a prototype integrated resource for functional and computational genomics". Genome Research. 9 (2): 195–209. doi:10.1101/gr.9.2.195. PMC 310711. PMID 10022985.
- ↑ Velculescu VE, Zhang L, Zhou W, Vogelstein J, Basrai MA, Bassett DE, Hieter P, Vogelstein B, Kinzler KW (January 1997). "Characterization of the yeast transcriptome". Cell. 88 (2): 243–51. doi:10.1016/S0092-8674(00)81845-0. PMID 9008165. S2CID 11430660.
- 1 2 3 4 Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (October 1995). "Serial analysis of gene expression". Science. 270 (5235): 484–7. Bibcode:1995Sci...270..484V. doi:10.1126/science.270.5235.484. PMID 7570003. S2CID 16281846.
- ↑ Audic S, Claverie JM (October 1997). "The significance of digital gene expression profiles". Genome Research. 7 (10): 986–95. doi:10.1101/gr.7.10.986. PMID 9331369.
- 1 2 3 4 5 6 Mantione KJ, Kream RM, Kuzelova H, Ptacek R, Raboch J, Samuel JM, Stefano GB (August 2014). "Comparing bioinformatic gene expression profiling methods: microarray and RNA-Seq". Medical Science Monitor Basic Research. 20: 138–42. doi:10.12659/MSMBR.892101. PMC 4152252. PMID 25149683.
- ↑ Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X (2014). "Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells". PLOS ONE. 9 (1): e78644. Bibcode:2014PLoSO...978644Z. doi:10.1371/journal.pone.0078644. PMC 3894192. PMID 24454679.
- 1 2 Hashimshony T, Wagner F, Sher N, Yanai I (September 2012). "CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification". Cell Reports. 2 (3): 666–73. doi:10.1016/j.celrep.2012.08.003. PMID 22939981.
- ↑ Stears RL, Getts RC, Gullans SR (August 2000). "A novel, sensitive detection system for high-density microarrays using dendrimer technology". Physiological Genomics. 3 (2): 93–9. doi:10.1152/physiolgenomics.2000.3.2.93. PMID 11015604.
- 1 2 3 4 5 6 Illumina (2011-07-11). "RNA-Seq Data Comparison with Gene Expression Microarrays" (PDF). European Pharmaceutical Review.
- 1 2 Black MB, Parks BB, Pluta L, Chu TM, Allen BC, Wolfinger RD, Thomas RS (February 2014). "Comparison of microarrays and RNA-seq for gene expression analyses of dose-response experiments". Toxicological Sciences. 137 (2): 385–403. doi:10.1093/toxsci/kft249. PMID 24194394.
- ↑ Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (September 2008). "RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays". Genome Research. 18 (9): 1509–17. doi:10.1101/gr.079558.108. PMC 2527709. PMID 18550803.
- ↑ SEQC/MAQC-III Consortium (September 2014). "A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium". Nature Biotechnology. 32 (9): 903–14. doi:10.1038/nbt.2957. PMC 4321899. PMID 25150838.
- ↑ Chen JJ, Hsueh HM, Delongchamp RR, Lin CJ, Tsai CA (October 2007). "Reproducibility of microarray data: a further analysis of microarray quality control (MAQC) data". BMC Bioinformatics. 8: 412. doi:10.1186/1471-2105-8-412. PMC 2204045. PMID 17961233.
- ↑ Larkin JE, Frank BC, Gavras H, Sultana R, Quackenbush J (May 2005). "Independence and reproducibility across microarray platforms". Nature Methods. 2 (5): 337–44. doi:10.1038/nmeth757. PMID 15846360. S2CID 16088782.
- 1 2 Nelson NJ (April 2001). "Microarrays have arrived: gene expression tool matures". Journal of the National Cancer Institute. 93 (7): 492–4. doi:10.1093/jnci/93.7.492. PMID 11287436.
- ↑ Schena M, Shalon D, Davis RW, Brown PO (October 1995). "Quantitative monitoring of gene expression patterns with a complementary DNA microarray". Science. 270 (5235): 467–70. Bibcode:1995Sci...270..467S. doi:10.1126/science.270.5235.467. PMID 7569999. S2CID 6720459.
- 1 2 Pozhitkov AE, Tautz D, Noble PA (June 2007). "Oligonucleotide microarrays: widely applied—poorly understood". Briefings in Functional Genomics & Proteomics. 6 (2): 141–8. doi:10.1093/bfgp/elm014. hdl:11858/00-001M-0000-000F-D7B3-3. PMID 17644526.
- 1 2 3 Heller MJ (2002). "DNA microarray technology: devices, systems, and applications". Annual Review of Biomedical Engineering. 4: 129–53. doi:10.1146/annurev.bioeng.4.020702.153438. PMID 12117754.
- ↑ McLachlan GJ, Do K, Ambroise C (2005). Analyzing Microarray Gene Expression Data. Hoboken: John Wiley & Sons. ISBN 978-0-471-72612-8.
- ↑ Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, Roth R, George D, Eletr S, Albrecht G, Vermaas E, Williams SR, Moon K, Burcham T, Pallas M, DuBridge RB, Kirchner J, Fearon K, Mao J, Corcoran K (June 2000). "Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays". Nature Biotechnology. 18 (6): 630–4. doi:10.1038/76469. PMID 10835600. S2CID 13884154.
- ↑ Meyers BC, Vu TH, Tej SS, Ghazal H, Matvienko M, Agrawal V, Ning J, Haudenschild CD (August 2004). "Analysis of the transcriptional complexity of Arabidopsis thaliana by massively parallel signature sequencing". Nature Biotechnology. 22 (8): 1006–11. doi:10.1038/nbt992. PMID 15247925. S2CID 15336496.
- 1 2 Bainbridge MN, Warren RL, Hirst M, Romanuik T, Zeng T, Go A, Delaney A, Griffith M, Hickenbotham M, Magrini V, Mardis ER, Sadar MD, Siddiqui AS, Marra MA, Jones SJ (September 2006). "Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach". BMC Genomics. 7: 246. doi:10.1186/1471-2164-7-246. PMC 1592491. PMID 17010196.
- ↑ Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (July 2008). "Mapping and quantifying mammalian transcriptomes by RNA-Seq". Nature Methods. 5 (7): 621–8. doi:10.1038/nmeth.1226. PMID 18516045. S2CID 205418589.
- ↑ Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bähler J (June 2008). "Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution". Nature. 453 (7199): 1239–43. Bibcode:2008Natur.453.1239W. doi:10.1038/nature07002. PMID 18488015. S2CID 205213499.
- ↑ Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O'Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML (August 2008). "A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome". Science. 321 (5891): 956–60. Bibcode:2008Sci...321..956S. doi:10.1126/science.1160342. PMID 18599741. S2CID 10013179.
- 1 2 Chomczynski P, Sacchi N (April 1987). "Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction". Analytical Biochemistry. 162 (1): 156–9. doi:10.1016/0003-2697(87)90021-2. PMID 2440339.
- 1 2 Chomczynski P, Sacchi N (2006). "The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on". Nature Protocols. 1 (2): 581–5. doi:10.1038/nprot.2006.83. PMID 17406285. S2CID 28653075.
- ↑ Grillo M, Margolis FL (September 1990). "Use of reverse transcriptase polymerase chain reaction to monitor expression of intronless genes". BioTechniques. 9 (3): 262, 264, 266–8. PMID 1699561.
- ↑ Bryant S, Manning DL (1998). "Isolation of messenger RNA". RNA Isolation and Characterization Protocols. Methods in Molecular Biology. Vol. 86. pp. 61–4. doi:10.1385/0-89603-494-1:61. ISBN 978-0-89603-494-5. PMID 9664454.
- ↑ Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM (June 2014). "Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling". BMC Genomics. 15 (1): 419. doi:10.1186/1471-2164-15-419. PMC 4070569. PMID 24888378.
- ↑ Some examples of environmental samples include: sea water, soil, or air.
- ↑ Close TJ, Wanamaker SI, Caldo RA, Turner SM, Ashlock DA, Dickerson JA, Wing RA, Muehlbauer GJ, Kleinhofs A, Wise RP (March 2004). "A new resource for cereal genomics: 22K barley GeneChip comes of age". Plant Physiology. 134 (3): 960–8. doi:10.1104/pp.103.034462. PMC 389919. PMID 15020760.
- 1 2 3 4 5 Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T (May 2017). "Transcriptomics technologies". PLOS Computational Biology. 13 (5): e1005457. Bibcode:2017PLSCB..13E5457L. doi:10.1371/journal.pcbi.1005457. PMC 5436640. PMID 28545146.
- 1 2 Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, Fukuda S, Sasaki D, Podhajska A, Harbers M, Kawai J, Carninci P, Hayashizaki Y (December 2003). "Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage". Proceedings of the National Academy of Sciences of the United States of America. 100 (26): 15776–81. Bibcode:2003PNAS..10015776S. doi:10.1073/pnas.2136655100. PMC 307644. PMID 14663149.
- ↑ Romanov V, Davidoff SN, Miles AR, Grainger DW, Gale BK, Brooks BD (March 2014). "A critical comparison of protein microarray fabrication technologies". The Analyst. 139 (6): 1303–26. Bibcode:2014Ana...139.1303R. doi:10.1039/c3an01577g. PMID 24479125.
- 1 2 Barbulovic-Nad I, Lucente M, Sun Y, Zhang M, Wheeler AR, Bussmann M (2006-10-01). "Bio-microarray fabrication techniques—a review". Critical Reviews in Biotechnology. 26 (4): 237–59. CiteSeerX 10.1.1.661.6833. doi:10.1080/07388550600978358. PMID 17095434. S2CID 13712888.
- ↑ Auburn RP, Kreil DP, Meadows LA, Fischer B, Matilla SS, Russell S (July 2005). "Robotic spotting of cDNA and oligonucleotide microarrays". Trends in Biotechnology. 23 (7): 374–9. doi:10.1016/j.tibtech.2005.04.002. PMID 15978318.
- ↑ Shalon D, Smith SJ, Brown PO (July 1996). "A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization". Genome Research. 6 (7): 639–45. doi:10.1101/gr.6.7.639. PMID 8796352.
- ↑ Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL (December 1996). "Expression monitoring by hybridization to high-density oligonucleotide arrays". Nature Biotechnology. 14 (13): 1675–80. doi:10.1038/nbt1296-1675. PMID 9634850. S2CID 35232673.
- ↑ Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (February 2003). "Summaries of Affymetrix GeneChip probe level data". Nucleic Acids Research. 31 (4): 15e–15. doi:10.1093/nar/gng015. PMC 150247. PMID 12582260.
- ↑ Selzer RR, Richmond TA, Pofahl NJ, Green RD, Eis PS, Nair P, Brothman AR, Stallings RL (November 2005). "Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH". Genes, Chromosomes & Cancer. 44 (3): 305–19. doi:10.1002/gcc.20243. PMID 16075461. S2CID 39437458.
- ↑ Svensson V, Vento-Tormo R, Teichmann SA (April 2018). "Exponential scaling of single-cell RNA-seq in the past decade". Nature Protocols. 13 (4): 599–604. doi:10.1038/nprot.2017.149. PMID 29494575. S2CID 3560001.
- ↑ Tachibana C (2015-08-18). "Transcriptomics today: Microarrays, RNA-seq, and more". Science. 349 (6247): 544. Bibcode:2015Sci...349..544T. doi:10.1126/science.opms.p1500095.
- 1 2 Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M (June 2008). "The transcriptional landscape of the yeast genome defined by RNA sequencing". Science. 320 (5881): 1344–9. Bibcode:2008Sci...320.1344N. doi:10.1126/science.1158441. PMC 2951732. PMID 18451266.
- ↑ Su Z, Fang H, Hong H, Shi L, Zhang W, Zhang W, Zhang Y, Dong Z, Lancashire LJ, Bessarabova M, Yang X, Ning B, Gong B, Meehan J, Xu J, Ge W, Perkins R, Fischer M, Tong W (December 2014). "An investigation of biomarkers derived from legacy microarray data for their utility in the RNA-seq era". Genome Biology. 15 (12): 523. doi:10.1186/s13059-014-0523-y. PMC 4290828. PMID 25633159.
- ↑ Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SS, Li C, Amamoto R, Peters DT, Turczyk BM, Marblestone AH, Inverso SA, Bernard A, Mali P, Rios X, Aach J, Church GM (March 2014). "Highly multiplexed subcellular RNA sequencing in situ". Science. 343 (6177): 1360–3. Bibcode:2014Sci...343.1360L. doi:10.1126/science.1250212. PMC 4140943. PMID 24578530.
- 1 2 Shendure J, Ji H (October 2008). "Next-generation DNA sequencing". Nature Biotechnology. 26 (10): 1135–45. doi:10.1038/nbt1486. PMID 18846087. S2CID 6384349.
- ↑ Lahens NF, Kavakli IH, Zhang R, Hayer K, Black MB, Dueck H, Pizarro A, Kim J, Irizarry R, Thomas RS, Grant GR, Hogenesch JB (June 2014). "IVT-seq reveals extreme bias in RNA sequencing". Genome Biology. 15 (6): R86. doi:10.1186/gb-2014-15-6-r86. PMC 4197826. PMID 24981968.
- 1 2 Knierim E, Lucke B, Schwarz JM, Schuelke M, Seelow D (2011). "Systematic comparison of three methods for fragmentation of long-range PCR products for next generation sequencing". PLOS ONE. 6 (11): e28240. Bibcode:2011PLoSO...628240K. doi:10.1371/journal.pone.0028240. PMC 3227650. PMID 22140562.
- ↑ Routh A, Head SR, Ordoukhanian P, Johnson JE (August 2015). "ClickSeq: Fragmentation-Free Next-Generation Sequencing via Click Ligation of Adaptors to Stochastically Terminated 3'-Azido cDNAs". Journal of Molecular Biology. 427 (16): 2610–6. doi:10.1016/j.jmb.2015.06.011. PMC 4523409. PMID 26116762.
- ↑ Parekh S, Ziegenhain C, Vieth B, Enard W, Hellmann I (May 2016). "The impact of amplification on differential expression analyses by RNA-seq". Scientific Reports. 6: 25533. Bibcode:2016NatSR...625533P. doi:10.1038/srep25533. PMC 4860583. PMID 27156886.
- ↑ Shanker S, Paulson A, Edenberg HJ, Peak A, Perera A, Alekseyev YO, Beckloff N, Bivens NJ, Donnelly R, Gillaspy AF, Grove D, Gu W, Jafari N, Kerley-Hamilton JS, Lyons RH, Tepper C, Nicolet CM (April 2015). "Evaluation of commercially available RNA amplification kits for RNA sequencing using very low input amounts of total RNA". Journal of Biomolecular Techniques. 26 (1): 4–18. doi:10.7171/jbt.15-2601-001. PMC 4310221. PMID 25649271.
- ↑ Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B (September 2011). "Synthetic spike-in standards for RNA-seq experiments". Genome Research. 21 (9): 1543–51. doi:10.1101/gr.121095.111. PMC 3166838. PMID 21816910.
- ↑ Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J (November 2011). "Counting absolute numbers of molecules using unique molecular identifiers". Nature Methods. 9 (1): 72–4. doi:10.1038/nmeth.1778. PMID 22101854. S2CID 39225091.
- ↑ Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, Lao K, Surani MA (May 2009). "mRNA-Seq whole-transcriptome analysis of a single cell". Nature Methods. 6 (5): 377–82. doi:10.1038/nmeth.1315. PMID 19349980. S2CID 16570747.
- ↑ Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S (February 2014). "Quantitative single-cell RNA-seq with unique molecular identifiers". Nature Methods. 11 (2): 163–6. doi:10.1038/nmeth.2772. PMID 24363023. S2CID 6765530.
- ↑ Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I (February 2014). "Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types". Science. 343 (6172): 776–9. Bibcode:2014Sci...343..776J. doi:10.1126/science.1247651. PMC 4412462. PMID 24531970.
- 1 2 Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A (September 2010). "Comprehensive comparative analysis of strand-specific RNA sequencing methods". Nature Methods. 7 (9): 709–15. doi:10.1038/nmeth.1491. PMC 3005310. PMID 20711195.
- ↑ Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (July 2012). "A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers". BMC Genomics. 13: 341. doi:10.1186/1471-2164-13-341. PMC 3431227. PMID 22827831.
- 1 2 Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M (2012). "Comparison of next-generation sequencing systems". Journal of Biomedicine & Biotechnology. 2012: 251364. doi:10.1155/2012/251364. PMC 3398667. PMID 22829749.
- ↑ "SRA". Retrieved 2016-10-06.The NCBI Sequence Read Archive (SRA) was searched using “RNA-Seq[Strategy]” and one of "LS454[Platform]”, “Illumina[platform]”, "ABI Solid[Platform]”, "Ion Torrent[Platform]”, "PacBio SMRT"[Platform]” to report the number of RNA-Seq runs deposited for each platform.
- ↑ Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ (May 2012). "Performance comparison of benchtop high-throughput sequencing platforms". Nature Biotechnology. 30 (5): 434–9. doi:10.1038/nbt.2198. PMID 22522955. S2CID 5300923.
- ↑ Goodwin S, McPherson JD, McCombie WR (May 2016). "Coming of age: ten years of next-generation sequencing technologies". Nature Reviews Genetics. 17 (6): 333–51. doi:10.1038/nrg.2016.49. PMC 10373632. PMID 27184599. S2CID 8295541.
- ↑ Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, Jordan M, Ciccone J, Serra S, Keenan J, Martin S, McNeill L, Wallace EJ, Jayasinghe L, Wright C, Blasco J, Young S, Brocklebank D, Juul S, Clarke J, Heron AJ, Turner DJ (March 2018). "Highly parallel direct RNA sequencing on an array of nanopores". Nature Methods. 15 (3): 201–206. doi:10.1038/nmeth.4577. PMID 29334379. S2CID 3589823.
- ↑ Loman NJ, Quick J, Simpson JT (August 2015). "A complete bacterial genome assembled de novo using only nanopore sequencing data". Nature Methods. 12 (8): 733–5. doi:10.1038/nmeth.3444. PMID 26076426. S2CID 15053702.
- ↑ Ozsolak F, Platt AR, Jones DR, Reifenberger JG, Sass LE, McInerney P, Thompson JF, Bowers J, Jarosz M, Milos PM (October 2009). "Direct RNA sequencing". Nature. 461 (7265): 814–8. Bibcode:2009Natur.461..814O. doi:10.1038/nature08390. PMID 19776739. S2CID 4426760.
- 1 2 Hart SN, Therneau TM, Zhang Y, Poland GA, Kocher JP (December 2013). "Calculating sample size estimates for RNA sequencing data". Journal of Computational Biology. 20 (12): 970–8. doi:10.1089/cmb.2012.0283. PMC 3842884. PMID 23961961.
- 1 2 3 Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A (January 2016). "A survey of best practices for RNA-seq data analysis". Genome Biology. 17: 13. doi:10.1186/s13059-016-0881-8. PMC 4728800. PMID 26813401.
- 1 2 Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, Mason CE, Socci ND, Betel D (2013). "Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data". Genome Biology. 14 (9): R95. doi:10.1186/gb-2013-14-9-r95. PMC 4054597. PMID 24020486.
- ↑ ENCODE Project Consortium; Aldred, Shelley F.; Collins, Patrick J.; Davis, Carrie A.; Doyle, Francis; Epstein, Charles B.; Frietze, Seth; Harrow, Jennifer; Kaul, Rajinder; Khatun, Jainab; Lajoie, Bryan R.; Landt, Stephen G.; Lee, Bum-Kyu; Pauli, Florencia; Rosenbloom, Kate R.; Sabo, Peter; Safi, Alexias; Sanyal, Amartya; Shoresh, Noam; Simon, Jeremy M.; Song, Lingyun; Altshuler, Robert C.; Birney, Ewan; Brown, James B.; Cheng, Chao; Djebali, Sarah; Dong, Xianjun; Dunham, Ian; Ernst, Jason; et al. (September 2012). "An integrated encyclopedia of DNA elements in the human genome". Nature. 489 (7414): 57–74. Bibcode:2012Natur.489...57T. doi:10.1038/nature11247. PMC 3439153. PMID 22955616.
- ↑ Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, et al. (January 2016). "ENCODE data at the ENCODE portal". Nucleic Acids Research. 44 (D1): D726–32. doi:10.1093/nar/gkv1160. PMC 4702836. PMID 26527727.
- ↑ "ENCODE: Encyclopedia of DNA Elements". encodeproject.org.
- 1 2 Thind AS, Monga I, Thakur PK, Kumari P, Dindhoria K, Krzak M, Ranson M, Ashford B (Nov 2021). "Demystifying emerging bulk RNA-Seq applications: the application and utility of bioinformatic methodology". Briefings in Bioinformatics. 22 (6). doi:10.1093/bib/bbab259. PMID 34329375.
- 1 2 Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (April 2015). "limma powers differential expression analyses for RNA-sequencing and microarray studies". Nucleic Acids Research. 43 (7): e47. doi:10.1093/nar/gkv007. PMC 4402510. PMID 25605792.
- 1 2 Robinson MD, McCarthy DJ, Smyth GK (January 2010). "edgeR: a Bioconductor package for differential expression analysis of digital gene expression data". Bioinformatics. 26 (1): 139–40. doi:10.1093/bioinformatics/btp616. PMC 2796818. PMID 19910308.
- 1 2 Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. (February 2015). "Orchestrating high-throughput genomic analysis with Bioconductor". Nature Methods. 12 (2): 115–21. doi:10.1038/nmeth.3252. PMC 4509590. PMID 25633503.
- ↑ Smyth, G. K. (2005). "Limma: Linear Models for Microarray Data". Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Statistics for Biology and Health. Springer, New York, NY. pp. 397–420. CiteSeerX 10.1.1.361.8519. doi:10.1007/0-387-29362-0_23. ISBN 9780387251462.
- ↑ Steve., Russell (2008). Microarray Technology in Practice. Meadows, Lisa A. Burlington: Elsevier. ISBN 9780080919768. OCLC 437246554.
- 1 2 Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (August 2013). "De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis". Nature Protocols. 8 (8): 1494–512. doi:10.1038/nprot.2013.084. PMC 3875132. PMID 23845962.
- 1 2 Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (March 2015). "StringTie enables improved reconstruction of a transcriptome from RNA-seq reads". Nature Biotechnology. 33 (3): 290–5. doi:10.1038/nbt.3122. PMC 4643835. PMID 25690850.
- ↑ Kodama Y, Shumway M, Leinonen R (January 2012). "The Sequence Read Archive: explosive growth of sequencing data". Nucleic Acids Research. 40 (Database issue): D54–6. doi:10.1093/nar/gkr854. PMC 3245110. PMID 22009675.
- 1 2 Edgar R, Domrachev M, Lash AE (January 2002). "Gene Expression Omnibus: NCBI gene expression and hybridization array data repository". Nucleic Acids Research. 30 (1): 207–10. doi:10.1093/nar/30.1.207. PMC 99122. PMID 11752295.
- ↑ Petrov A, Shams S (2004-11-01). "Microarray Image Processing and Quality Control". Journal of VLSI Signal Processing Systems for Signal, Image and Video Technology. 38 (3): 211–226. doi:10.1023/B:VLSI.0000042488.08307.ad. S2CID 31598448.
- ↑ Petrov A, Shams S (2004). "Microarray Image Processing and Quality Control". The Journal of VLSI Signal Processing-Systems for Signal, Image, and Video Technology. 38 (3): 211–226. doi:10.1023/B:VLSI.0000042488.08307.ad. S2CID 31598448.
- ↑ Kwon YM, Ricke S (2011). High-Throughput Next Generation Sequencing. Methods in Molecular Biology. Vol. 733. SpringerLink. doi:10.1007/978-1-61779-089-8. ISBN 978-1-61779-088-1. S2CID 3684245.
- ↑ Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, Ishikawa S, Linak MC, Hirai A, Takahashi H, Altaf-Ul-Amin M, Ogasawara N, Kanaya S (July 2011). "Sequence-specific error profile of Illumina sequencers". Nucleic Acids Research. 39 (13): e90. doi:10.1093/nar/gkr344. PMC 3141275. PMID 21576222.
- ↑ Van Verk MC, Hickman R, Pieterse CM, Van Wees SC (April 2013). "RNA-Seq: revelation of the messengers". Trends in Plant Science. 18 (4): 175–9. doi:10.1016/j.tplants.2013.02.001. hdl:1874/309456. PMID 23481128. S2CID 205453732.
- ↑ Andrews S (2010). "FastQC: A Quality Control tool for High Throughput Sequence Data". Babraham Bioinformatics. Retrieved 2017-05-23.
- ↑ Lo CC, Chain PS (November 2014). "Rapid evaluation and quality control of next generation sequencing data with FaQCs". BMC Bioinformatics. 15 (1): 366. doi:10.1186/s12859-014-0366-2. PMC 4246454. PMID 25408143.
- 1 2 3 Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (January 2013). "Differential analysis of gene regulation at transcript resolution with RNA-seq". Nature Biotechnology. 31 (1): 46–53. doi:10.1038/nbt.2450. PMC 3869392. PMID 23222703.
- 1 2 Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, Zhou X, Lam TW, Li Y, Xu X, Wong GK, Wang J (June 2014). "SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads". Bioinformatics. 30 (12): 1660–6. arXiv:1305.6760. doi:10.1093/bioinformatics/btu077. PMID 24532719. S2CID 5152689.
- ↑ Siadjeu, Christian; Mayland-Quellhorst, Eike; Pande, Shruti; Laubinger, Sascha; Albach, Dirk C. (2021). "Transcriptome Sequence Reveals Candidate Genes Involving in the Post-Harvest Hardening of Trifoliate Yam Dioscorea dumetorum". Plants. 10 (4): 787. doi:10.3390/plants10040787. PMC 8074181. PMID 33923758.
- ↑ HTS Mappers. http://www.ebi.ac.uk/~nf/hts_mappers/
- ↑ Fonseca NA, Rung J, Brazma A, Marioni JC (December 2012). "Tools for mapping high-throughput sequencing data". Bioinformatics. 28 (24): 3169–77. doi:10.1093/bioinformatics/bts605. PMID 23060614.
- ↑ Trapnell C, Pachter L, Salzberg SL (May 2009). "TopHat: discovering splice junctions with RNA-Seq". Bioinformatics. 25 (9): 1105–11. doi:10.1093/bioinformatics/btp120. PMC 2672628. PMID 19289445.
- 1 2 Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (May 2010). "Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation". Nature Biotechnology. 28 (5): 511–5. doi:10.1038/nbt.1621. PMC 3146043. PMID 20436464.
- ↑ Miller JR, Koren S, Sutton G (June 2010). "Assembly algorithms for next-generation sequencing data". Genomics. 95 (6): 315–27. doi:10.1016/j.ygeno.2010.03.001. PMC 2874646. PMID 20211242.
- ↑ O'Neil ST, Emrich SJ (July 2013). "Assessing De Novo transcriptome assembly metrics for consistency and utility". BMC Genomics. 14: 465. doi:10.1186/1471-2164-14-465. PMC 3733778. PMID 23837739.
- ↑ Smith-Unna R, Boursnell C, Patro R, Hibberd JM, Kelly S (August 2016). "TransRate: reference-free quality assessment of de novo transcriptome assemblies". Genome Research. 26 (8): 1134–44. doi:10.1101/gr.196469.115. PMC 4971766. PMID 27252236.
- ↑ Li B, Fillmore N, Bai Y, Collins M, Thomson JA, Stewart R, Dewey CN (December 2014). "Evaluation of de novo transcriptome assemblies from RNA-Seq data". Genome Biology. 15 (12): 553. doi:10.1186/s13059-014-0553-5. PMC 4298084. PMID 25608678.
- ↑ Zerbino DR, Birney E (May 2008). "Velvet: algorithms for de novo short read assembly using de Bruijn graphs". Genome Research. 18 (5): 821–9. doi:10.1101/gr.074492.107. PMC 2336801. PMID 18349386.
- ↑ Schulz MH, Zerbino DR, Vingron M, Birney E (April 2012). "Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels". Bioinformatics. 28 (8): 1086–92. doi:10.1093/bioinformatics/bts094. PMC 3324515. PMID 22368243.
- ↑ Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, et al. (November 2010). "De novo assembly and analysis of RNA-seq data". Nature Methods. 7 (11): 909–12. doi:10.1038/nmeth.1517. hdl:1885/51040. PMID 20935650. S2CID 1034682.
- 1 2 Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (May 2011). "Full-length transcriptome assembly from RNA-Seq data without a reference genome". Nature Biotechnology. 29 (7): 644–52. doi:10.1038/nbt.1883. PMC 3571712. PMID 21572440.
- ↑ Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WE, Wetter T, Suhai S (June 2004). "Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs". Genome Research. 14 (6): 1147–59. doi:10.1101/gr.1917404. PMC 419793. PMID 15140833.
- ↑ Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. (September 2005). "Genome sequencing in microfabricated high-density picolitre reactors". Nature. 437 (7057): 376–80. Bibcode:2005Natur.437..376M. doi:10.1038/nature03959. PMC 1464427. PMID 16056220.
- ↑ Kumar S, Blaxter ML (October 2010). "Comparing de novo assemblers for 454 transcriptome data". BMC Genomics. 11: 571. doi:10.1186/1471-2164-11-571. PMC 3091720. PMID 20950480.
- ↑ Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (May 2012). "SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing". Journal of Computational Biology. 19 (5): 455–77. doi:10.1089/cmb.2012.0021. PMC 3342519. PMID 22506599.
- ↑ Li B, Dewey CN (August 2011). "RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome". BMC Bioinformatics. 12: 323. doi:10.1186/1471-2105-12-323. PMC 3163565. PMID 21816040.
- ↑ Kovaka, Sam; Zimin, Aleksey V.; Pertea, Geo M.; Razaghi, Roham; Salzberg, Steven L.; Pertea, Mihaela (2019-07-08). "Transcriptome assembly from long-read RNA-seq alignments with StringTie2". bioRxiv: 694554. doi:10.1101/694554. Retrieved 27 August 2019.
- ↑ Gehlenborg N, O'Donoghue SI, Baliga NS, Goesmann A, Hibbs MA, Kitano H, Kohlbacher O, Neuweger H, Schneider R, Tenenbaum D, Gavin AC (March 2010). "Visualization of omics data for systems biology". Nature Methods. 7 (3 Suppl): S56–68. doi:10.1038/nmeth.1436. PMID 20195258. S2CID 205419270.
- ↑ Anders S, Pyl PT, Huber W (January 2015). "HTSeq—a Python framework to work with high-throughput sequencing data". Bioinformatics. 31 (2): 166–9. doi:10.1093/bioinformatics/btu638. PMC 4287950. PMID 25260700.
- ↑ Bray NL, Pimentel H, Melsted P, Pachter L (May 2016). "Near-optimal probabilistic RNA-seq quantification". Nature Biotechnology. 34 (5): 525–7. doi:10.1038/nbt.3519. PMID 27043002. S2CID 205282743.
- ↑ Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (August 2009). "The Sequence Alignment/Map format and SAMtools". Bioinformatics. 25 (16): 2078–9. doi:10.1093/bioinformatics/btp352. PMC 2723002. PMID 19505943.
- ↑ Love MI, Huber W, Anders S (2014). "Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2". Genome Biology. 15 (12): 550. doi:10.1186/s13059-014-0550-8. PMC 4302049. PMID 25516281.
- ↑ Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT (March 2015). "Ballgown bridges the gap between transcriptome assembly and expression analysis". Nature Biotechnology. 33 (3): 243–6. doi:10.1038/nbt.3172. PMC 4792117. PMID 25748911.
- ↑ Fang Z, Cui X (May 2011). "Design and validation issues in RNA-seq experiments". Briefings in Bioinformatics. 12 (3): 280–7. doi:10.1093/bib/bbr004. PMID 21498551.
- ↑ Ramsköld D, Wang ET, Burge CB, Sandberg R (December 2009). "An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data". PLOS Computational Biology. 5 (12): e1000598. Bibcode:2009PLSCB...5E0598R. doi:10.1371/journal.pcbi.1000598. PMC 2781110. PMID 20011106.
- ↑ Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (June 2002). "Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes". Genome Biology. 3 (7): RESEARCH0034. doi:10.1186/gb-2002-3-7-research0034. PMC 126239. PMID 12184808.
- ↑ Core LJ, Waterfall JJ, Lis JT (December 2008). "Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters". Science. 322 (5909): 1845–8. Bibcode:2008Sci...322.1845C. doi:10.1126/science.1162228. PMC 2833333. PMID 19056941.
- ↑ Camarena L, Bruno V, Euskirchen G, Poggio S, Snyder M (April 2010). "Molecular mechanisms of ethanol-induced pathogenesis revealed by RNA-sequencing". PLOS Pathogens. 6 (4): e1000834. doi:10.1371/journal.ppat.1000834. PMC 2848557. PMID 20368969.
- 1 2 Govind G, Harshavardhan VT, ThammeGowda HV, Patricia JK, Kalaiarasi PJ, Dhanalakshmi R, Iyer DR, Senthil Kumar M, Muthappa SK, Sreenivasulu N, Nese S, Udayakumar M, Makarla UK (June 2009). "Identification and functional validation of a unique set of drought induced genes preferentially expressed in response to gradual water stress in peanut". Molecular Genetics and Genomics. 281 (6): 591–605. doi:10.1007/s00438-009-0432-z. PMC 2757612. PMID 19224247.
- ↑ Tavassoly, Iman; Goldfarb, Joseph; Iyengar, Ravi (2018-10-04). "Systems biology primer: the basic methods and approaches". Essays in Biochemistry. 62 (4): 487–500. doi:10.1042/EBC20180003. ISSN 0071-1365. PMID 30287586. S2CID 52922135.
- ↑ Costa V, Aprile M, Esposito R, Ciccodicola A (February 2013). "RNA-Seq and human complex diseases: recent accomplishments and future perspectives". European Journal of Human Genetics. 21 (2): 134–42. doi:10.1038/ejhg.2012.129. PMC 3548270. PMID 22739340.
- ↑ Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, Gerstein M (February 2016). "Role of non-coding sequence variants in cancer". Nature Reviews Genetics. 17 (2): 93–108. doi:10.1038/nrg.2015.17. PMID 26781813. S2CID 14433306.
- ↑ Slotkin RK, Martienssen R (April 2007). "Transposable elements and the epigenetic regulation of the genome". Nature Reviews Genetics. 8 (4): 272–85. doi:10.1038/nrg2072. PMID 17363976. S2CID 9719784.
- ↑ Proserpio V, Mahata B (February 2016). "Single-cell technologies to study the immune system". Immunology. 147 (2): 133–40. doi:10.1111/imm.12553. PMC 4717243. PMID 26551575.
- 1 2 Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW (May 2016). "Translating RNA sequencing into clinical diagnostics: opportunities and challenges". Nature Reviews Genetics. 17 (5): 257–71. doi:10.1038/nrg.2016.10. PMC 7097555. PMID 26996076.
- ↑ Wu HJ, Wang AH, Jennings MP (February 2008). "Discovery of virulence factors of pathogenic bacteria" (PDF). Current Opinion in Chemical Biology. 12 (1): 93–101. doi:10.1016/j.cbpa.2008.01.023. PMID 18284925.
- ↑ Suzuki S, Horinouchi T, Furusawa C (December 2014). "Prediction of antibiotic resistance by gene expression profiles". Nature Communications. 5: 5792. Bibcode:2014NatCo...5.5792S. doi:10.1038/ncomms6792. PMC 4351646. PMID 25517437.
- ↑ Westermann AJ, Gorski SA, Vogel J (September 2012). "Dual RNA-seq of pathogen and host" (PDF). Nature Reviews. Microbiology. 10 (9): 618–30. doi:10.1038/nrmicro2852. PMID 22890146. S2CID 205498287.
- ↑ Durmuş S, Çakır T, Özgür A, Guthke R (2015). "A review on computational systems biology of pathogen-host interactions". Frontiers in Microbiology. 6: 235. doi:10.3389/fmicb.2015.00235. PMC 4391036. PMID 25914674.
- 1 2 Garg R, Shankar R, Thakkar B, Kudapa H, Krishnamurthy L, Mantri N, Varshney RK, Bhatia S, Jain M (January 2016). "Transcriptome analyses reveal genotype- and developmental stage-specific molecular responses to drought and salinity stresses in chickpea". Scientific Reports. 6: 19228. Bibcode:2016NatSR...619228G. doi:10.1038/srep19228. PMC 4725360. PMID 26759178.
- ↑ García-Sánchez S, Aubert S, Iraqui I, Janbon G, Ghigo JM, d'Enfert C (April 2004). "Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns". Eukaryotic Cell. 3 (2): 536–45. doi:10.1128/EC.3.2.536-545.2004. PMC 387656. PMID 15075282.
- ↑ Rich SM, Leendertz FH, Xu G, LeBreton M, Djoko CF, Aminake MN, Takang EE, Diffo JL, Pike BL, Rosenthal BM, Formenty P, Boesch C, Ayala FJ, Wolfe ND (September 2009). "The origin of malignant malaria". Proceedings of the National Academy of Sciences of the United States of America. 106 (35): 14902–7. Bibcode:2009PNAS..10614902R. doi:10.1073/pnas.0907740106. PMC 2720412. PMID 19666593.
- ↑ Mok S, Ashley EA, Ferreira PE, Zhu L, Lin Z, Yeo T, et al. (January 2015). "Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance". Science. 347 (6220): 431–5. Bibcode:2015Sci...347..431M. doi:10.1126/science.1260403. PMC 5642863. PMID 25502316.
- 1 2 3 4 Page, Tessa M.; Lawley, Jonathan W. (2022). "The Next Generation Is Here: A Review of Transcriptomic Approaches in Marine Ecology". Frontiers in Marine Science. 9. doi:10.3389/fmars.2022.757921. ISSN 2296-7745.
- ↑ Verbruggen N, Hermans C, Schat H (March 2009). "Molecular mechanisms of metal hyperaccumulation in plants" (PDF). The New Phytologist. 181 (4): 759–76. doi:10.1111/j.1469-8137.2008.02748.x. PMID 19192189.
- ↑ Li Z, Zhang Z, Yan P, Huang S, Fei Z, Lin K (November 2011). "RNA-Seq improves annotation of protein-coding genes in the cucumber genome". BMC Genomics. 12: 540. doi:10.1186/1471-2164-12-540. PMC 3219749. PMID 22047402.
- ↑ Hobbs M, Pavasovic A, King AG, Prentis PJ, Eldridge MD, Chen Z, Colgan DJ, Polkinghorne A, Wilkins MR, Flanagan C, Gillett A, Hanger J, Johnson RN, Timms P (September 2014). "A transcriptome resource for the koala (Phascolarctos cinereus): insights into koala retrovirus transcription and sequence diversity". BMC Genomics. 15 (1): 786. doi:10.1186/1471-2164-15-786. PMC 4247155. PMID 25214207.
- ↑ Howe GT, Yu J, Knaus B, Cronn R, Kolpak S, Dolan P, Lorenz WW, Dean JF (February 2013). "A SNP resource for Douglas-fir: de novo transcriptome assembly and SNP detection and validation". BMC Genomics. 14: 137. doi:10.1186/1471-2164-14-137. PMC 3673906. PMID 23445355.
- ↑ McGrath LL, Vollmer SV, Kaluziak ST, Ayers J (January 2016). "De novo transcriptome assembly for the lobster Homarus americanus and characterization of differential gene expression across nervous system tissues". BMC Genomics. 17: 63. doi:10.1186/s12864-016-2373-3. PMC 4715275. PMID 26772543.
- 1 2 Meyer DH, Schumacher B (2020). "BiT age: A transcriptome-based aging clock near the theoretical limit of accuracy". Aging Cell. 20 (3): e13320. doi:10.1111/acel.13320. PMC 7963339. PMID 33656257.
- ↑ Fleischer JG, Schulte R, Tsai HH, Tyagi S, Ibarra A, Shokhirev MN, Navlakha S (2018). "Predicting age from the transcriptome of human dermal fibroblasts". Genome Biology. 19 (1): 221. doi:10.1186/s13059-018-1599-6. PMC 6300908. PMID 30567591.
- ↑ Noller HF (1991). "Ribosomal RNA and translation". Annual Review of Biochemistry. 60: 191–227. doi:10.1146/annurev.bi.60.070191.001203. PMID 1883196.
- ↑ Christov CP, Gardiner TJ, Szüts D, Krude T (September 2006). "Functional requirement of noncoding Y RNAs for human chromosomal DNA replication". Molecular and Cellular Biology. 26 (18): 6993–7004. doi:10.1128/MCB.01060-06. PMC 1592862. PMID 16943439.
- ↑ Kishore S, Stamm S (January 2006). "The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C". Science. 311 (5758): 230–2. Bibcode:2006Sci...311..230K. doi:10.1126/science.1118265. PMID 16357227. S2CID 44527461.
- ↑ Hüttenhofer A, Schattner P, Polacek N (May 2005). "Non-coding RNAs: hope or hype?". Trends in Genetics. 21 (5): 289–97. doi:10.1016/j.tig.2005.03.007. PMID 15851066.
- ↑ Esteller M (November 2011). "Non-coding RNAs in human disease". Nature Reviews Genetics. 12 (12): 861–74. doi:10.1038/nrg3074. PMID 22094949. S2CID 13036469.
- ↑ "Gene Expression Omnibus". www.ncbi.nlm.nih.gov. Retrieved 2018-03-26.
- 1 2 Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M (December 2001). "Minimum information about a microarray experiment (MIAME)-toward standards for microarray data". Nature Genetics. 29 (4): 365–71. doi:10.1038/ng1201-365. PMID 11726920. S2CID 6994467.
- 1 2 Brazma A (May 2009). "Minimum Information About a Microarray Experiment (MIAME)--successes, failures, challenges". TheScientificWorldJournal. 9: 420–3. doi:10.1100/tsw.2009.57. PMC 5823224. PMID 19484163.
- ↑ Kolesnikov N, Hastings E, Keays M, Melnichuk O, Tang YA, Williams E, Dylag M, Kurbatova N, Brandizi M, Burdett T, Megy K, Pilicheva E, Rustici G, Tikhonov A, Parkinson H, Petryszak R, Sarkans U, Brazma A (January 2015). "ArrayExpress update—simplifying data submissions". Nucleic Acids Research. 43 (Database issue): D1113–6. doi:10.1093/nar/gku1057. PMC 4383899. PMID 25361974.
- ↑ Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, Füllgrabe A, Fuentes AM, Jupp S, Koskinen S, Mannion O, Huerta L, Megy K, Snow C, Williams E, Barzine M, Hastings E, Weisser H, Wright J, Jaiswal P, Huber W, Choudhary J, Parkinson HE, Brazma A (January 2016). "Expression Atlas update—an integrated database of gene and protein expression in humans, animals and plants". Nucleic Acids Research. 44 (D1): D746–52. doi:10.1093/nar/gkv1045. PMC 4702781. PMID 26481351.
- ↑ Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, Widmayer P, Gruissem W, Zimmermann P (2008). "Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes". Advances in Bioinformatics. 2008: 420747. doi:10.1155/2008/420747. PMC 2777001. PMID 19956698.
- ↑ Mitsuhashi N, Fujieda K, Tamura T, Kawamoto S, Takagi T, Okubo K (January 2009). "BodyParts3D: 3D structure database for anatomical concepts". Nucleic Acids Research. 37 (Database issue): D782–5. doi:10.1093/nar/gkn613. PMC 2686534. PMID 18835852.
- ↑ Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, Li Z, Bu D, Sun N, Zhang MQ, Chen R (January 2016). "NONCODE 2016: an informative and valuable data source of long non-coding RNAs". Nucleic Acids Research. 44 (D1): D203–8. doi:10.1093/nar/gkv1252. PMC 4702886. PMID 26586799.
Notes
Further reading
- Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T (May 2017). "Transcriptomics technologies". PLOS Computational Biology. 13 (5): e1005457. Bibcode:2017PLSCB..13E5457L. doi:10.1371/journal.pcbi.1005457. PMC 5436640. PMID 28545146.
- Comparative Transcriptomics Analysis in Reference Module in Life Sciences
- Software used in transcriptomics: