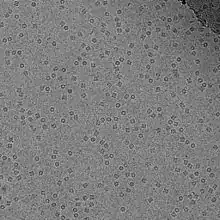
Transmission electron cryomicroscopy (CryoTEM), commonly known as cryo-EM, is a form of cryogenic electron microscopy, more specifically a type of transmission electron microscopy (TEM) where the sample is studied at cryogenic temperatures (generally liquid-nitrogen temperatures).[1] Cryo-EM, specifically 3-dimensional electron microscopy (3DEM), is gaining popularity in structural biology.[2]
The utility of transmission electron cryomicroscopy stems from the fact that it allows the observation of specimens that have not been stained or fixed in any way, showing them in their native environment. This is in contrast to X-ray crystallography, which requires crystallizing the specimen, which can be difficult, and placing them in non-physiological environments, which can occasionally lead to functionally irrelevant conformational changes.
Advances in electron detector technology, particularly DED (Direct Electron Detectors) as well as more powerful software imaging algorithms have allowed for the determination of macromolecular structures at near-atomic resolution.[3] Imaged macromolecules include viruses, ribosomes, mitochondria, ion channels, and enzyme complexes. Starting in 2018, cryo-EM could applied to structures as small as hemoglobin (64 kDa)[4] and with resolutions up to 1.8 Å.[5] In 2019, cryo-EM structures represented 2.5% of structures deposited in the Protein Data Bank,[6] and this number continues to grow.[7] An application of cryo-EM is cryo-electron tomography (cryo-ET), where a 3D reconstruction of the sample is created from tilted 2D images.
Development
The original rationale for CryoTEM was as a means to fight radiation damage for biological specimens. The amount of radiation required to collect an image of a specimen in the electron microscope is high enough to be a potential source of specimen damage for delicate structures. In addition, the high vacuum required on the column of an electron microscope makes the environment for the sample quite harsh.
The problem of the vacuum was partially solved by the introduction of negative stains but even with negative stains biological samples are prone to structural collapse upon dehydration of the specimen. Embedding the samples in ice below the sublimation temperature was a possibility that was contemplated early on, but water tends to arrange into a crystalline lattice of lower density upon freezing and this can destroy the structure of anything that is embedded in it.
In the early 1980s, several groups studying solid state physics were attempting to produce vitreous ice by different means, such as high pressure freezing or flash freezing. In a seminal paper in 1984, the group led by Jacques Dubochet at the European Molecular Biology Laboratory showed images of adenovirus embedded in a vitrified layer of water.[8] This paper is generally considered to mark the origin of Cryo-EM, and the technique has been developed to the point of becoming routine at numerous laboratories throughout the world.
The energy of the electrons used for imaging (80–300 kV) is high enough that covalent bonds can be broken. When imaging specimens are vulnerable to radiation damage, it is necessary to limit the electron exposure used to acquire the image. These low exposures require that the images of thousands or even millions of identical frozen molecules be selected, aligned, and averaged to obtain high-resolution maps, using specialized software. A significant improvement in structural features was achieved in 2012 by the introduction of direct electron detectors and better computational algorithms.[1][2]
In 2015, Bridget Carragher and colleagues at the Scripps National Resource for Automated Molecular Microscopy used techniques she and Clint Potter developed to determine the first cryo-EM structure with a resolution finer than 3 Å, thereby elevating CryoTEM as a tool comparable to and potentially superior to traditional x-ray crystallography techniques.[9][10] Since then, higher resolutions have been achieved, including a 2.2 Å structure of bacterial enzyme β-galactosidase in 2015[11] and a 1.8 Å structure of glutamate dehydrogenase in 2016.[12] Cryo-EM has also been used to determine the structure of various viruses, including the Zika virus,[13] and has been applied to large complexes such as the spliceosome.[14] In 2017, the Nobel Prize in Chemistry was awarded jointly to Jacques Dubochet, Joachim Frank and Richard Henderson, "for developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution".[15]
Biological specimens
Thin film
The biological material is spread on an electron microscopy grid and is preserved in a frozen-hydrated state by rapid freezing, usually in liquid ethane near liquid nitrogen temperature. By maintaining specimens at liquid nitrogen temperature or colder, they can be introduced into the high-vacuum of the electron microscope column. Most biological specimens are extremely radiosensitive, so they must be imaged with low-dose techniques (usefully, the low temperature of transmission electron cryomicroscopy provides an additional protective factor against radiation damage).
Consequently, the images are extremely noisy. For some biological systems it is possible to average images to increase the signal-to-noise ratio and retrieve high-resolution information about the specimen using the technique known as single particle analysis. This approach in general requires that the things being averaged are identical, although some limited conformational heterogeneity can now be studied (e.g. ribosome). Three-dimensional reconstructions from CryoTEM images of protein complexes and viruses have been solved to sub-nanometer or near-atomic resolution, allowing new insights into the structure and biology of these large assemblies.
Analysis of ordered arrays of protein, such as 2-D crystals of transmembrane proteins or helical arrays of proteins, also allows a kind of averaging which can provide high-resolution information about the specimen. This technique is called electron crystallography.
Vitreous sections
The thin film method is limited to thin specimens (typically < 500 nm) because the electrons cannot cross thicker samples without multiple scattering events. Thicker specimens can be vitrified by plunge freezing (cryofixation) in ethane (up to tens of μm in thickness) or more commonly by high pressure freezing (up to hundreds of μm). They can then be cut in thin sections (40 to 200 nm thick) with a diamond knife in a cryoultramicrotome at temperatures lower than −135 °C (devitrification temperature). The sections are collected on an electron microscope grid and are imaged in the same manner as specimen vitrified in thin film. This technique is called transmission electron cryomicroscopy of vitreous sections (CEMOVIS) or transmission electron cryomicroscopy of frozen-hydrated sections.
Material specimens
In addition to allowing vitrified biological samples to be imaged, CryoTEM can also be used to image material specimens that are too volatile in vacuum to image using standard, room temperature electron microscopy. For example, vitrified sections of liquid-solid interfaces can be extracted for analysis by CryoTEM,[16] and sulfur, which is prone to sublimation in the vacuum of electron microscopes, can be stabilized and imaged in CryoTEM.[17]
Image processing in cryo-TEM
Even though in the majority of approaches in electron microscopy one tries to get the best resolution image of the material, it is not always the case in cryo-TEM. Besides all the benefits of high resolution images, the signal to noise ratio remains the main hurdle that prevents assigning orientation to each particle. For example, in macromolecule complexes, there are several different structures that are being projected from 3D to 2D during imaging and if they are not distinguished the result of image processing will be a blur. That is why the probabilistic approaches become more powerful in this type of investigation.[18] There are two popular approaches that are widely used nowadays in cryo-EM image processing, the maximum likelihood approach that was discovered in 1998[19] and relatively recently adapted Bayesian approach.[20]
The maximum likelihood estimation approach comes to this field from the statistics. Here, all the possible orientations of particles are summed up to get the resulting probability distribution. We can compare this to a typical least square estimation where particles get exact orientations per image.[21] This way, the particles in the sample get "fuzzy" orientations after calculations, weighted by corresponding probabilities. The whole process is iterative and with each next iteration the model gets better. The good conditions for making the model that closely represent the real structure is when the data does not have too much noise and the particles do not have any preferential direction. The main downside of maximum likelihood approach is that the result depends on the initial guess and model optimization can sometimes get stuck at local minimum.[22]
The Bayesian approach that is now being used in cryo-TEM is empirical by nature. This means that the distribution of particles is based on the original dataset. Similarly, in the usual Bayesian method there is a fixed prior probability that is changed after the data is observed. The main difference from the maximum likelihood estimation lies in special reconstruction term that helps smoothing the resulting maps while also decreasing the noise during reconstruction.[21] The smoothing of the maps occurs through assuming prior probability to be a Gaussian distribution and analyzing the data in the Fourier space. Since the connection between the prior knowledge and the dataset is established, there is less chance for human factor errors which potentially increases the objectivity of image reconstruction.[20]
With emerging new methods of cryo-TEM imaging and image reconstruction the new software solutions appear that help to automate the process. After the empirical Bayesian approach have been implemented in the open source computer program RELION (REgularized LIkelihood OptimizatioN) for 3D reconstruction,[23][24] the program became widespread in the cryo-TEM field. It offers a range of corrections that improve the resolution of reconstructed images, allows implementing versatile scripts using python language and executes the usual tasks of 2D/3D model classifications or creating de novo models.[25][26]
Techniques
A variety of techniques can be used in CryoTEM.[27] Popular techniques include:
- Electron crystallography
- Analysis of two-dimensional crystals
- Analysis of helical filaments or tubes
- Microcrystal Electron Diffraction (MicroED)[28][29][30][31]
- Single particle analysis (SPA)
- Electron cryotomography (cryoET)
See also
References
- 1 2 Kühlbrandt W (August 2014). "Cryo-EM enters a new era". eLife. 3: e03678. doi:10.7554/elife.03678. PMC 4131193. PMID 25122623.
- 1 2 Callaway E (September 2015). "The revolution will not be crystallized: a new method sweeps through structural biology". Nature. 525 (7568): 172–4. Bibcode:2015Natur.525..172C. doi:10.1038/525172a. PMID 26354465.
- ↑ Murata K, Wolf M (Feb 2018). "Cryo-electron microscopy for structural analysis of dynamic biological macromolecules". Biochimica et Biophysica Acta (BBA) - General Subjects. 1862 (2): 324–334. doi:10.1016/j.bbagen.2017.07.020. PMID 28756276.
- ↑ Khoshouei M, Radjainia M, Baumeister W, Danev R (June 2017). "Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate". Nature Communications. 8: 16099. Bibcode:2017NatCo...816099K. doi:10.1038/ncomms16099. PMC 5497076. PMID 28665412.
- ↑ Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JL, Subramaniam S (June 2016). "Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery". Cell. 165 (7): 1698–1707. doi:10.1016/j.cell.2016.05.040. PMC 4931924. PMID 27238019.
- ↑ "PDB Data Distribution by Experimental Method and Molecular Type". www.rcsb.org. Retrieved 2019-12-03.
- ↑ "PDB Statistics: Growth of Structures from 3DEM Experiments Released per Year". www.rcsb.org. Retrieved 2018-12-22.
- ↑ Adrian M, Dubochet J, Lepault J, McDowall AW (1984). "Cryo-electron microscopy of viruses". Nature. 308 (5954): 32–6. Bibcode:1984Natur.308...32A. doi:10.1038/308032a0. PMID 6322001. S2CID 4319199.
- ↑ Dellisanti, Cosma (2015). "A barrier-breaking resolution". Nature Structural & Molecular Biology. 22 (5): 361. doi:10.1038/nsmb.3025. S2CID 12198387.
- ↑ Campbell MG, Veesler D, Cheng A, Potter CS, Carragher B (March 2015). "2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy". eLife. 4. doi:10.7554/eLife.06380. PMC 4391500. PMID 25760083.
- ↑ Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JL, Subramaniam S (June 2015). "2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor". Science. 348 (6239): 1147–51. Bibcode:2015Sci...348.1147B. doi:10.1126/science.aab1576. PMC 6512338. PMID 25953817.
- ↑ Vonck J, Mills DJ (October 2017). "Advances in high-resolution cryo-EM of oligomeric enzymes". Current Opinion in Structural Biology. 46: 48–54. doi:10.1016/j.sbi.2017.05.016. PMID 28624735.
- ↑ Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ (April 2016). "The 3.8 Å resolution cryo-EM structure of Zika virus". Science. 352 (6284): 467–70. Bibcode:2016Sci...352..467S. doi:10.1126/science.aaf5316. PMC 4845755. PMID 27033547.
- ↑ Cheng Y (August 2018). "Single-particle cryo-EM-How did it get here and where will it go". Science. 361 (6405): 876–880. Bibcode:2018Sci...361..876C. doi:10.1126/science.aat4346. PMC 6460916. PMID 30166484.
- ↑ "The 2017 Nobel Prize in Chemistry – Press Release". www.nobelprize.org. 4 October 2017. Retrieved 4 October 2017.
- ↑ Zachman MJ, Asenath-Smith E, Estroff LA, Kourkoutis LF (December 2016). "Site-Specific Preparation of Intact Solid-Liquid Interfaces by Label-Free In Situ Localization and Cryo-Focused Ion Beam Lift-Out". Microscopy and Microanalysis. 22 (6): 1338–1349. Bibcode:2016MiMic..22.1338Z. doi:10.1017/S1431927616011892. PMID 27869059.
- ↑ Levin BD, Zachman MJ, Werner JG, Sahore R, Nguyen KX, Han Y, Xie B, Ma L, Archer LA, Giannelis EP, Wiesner U, Kourkoutis LF, Muller DA (February 2017). "Characterization of Sulfur and Nanostructured Sulfur Battery Cathodes in Electron Microscopy Without Sublimation Artifacts". Microscopy and Microanalysis. 23 (1): 155–162. Bibcode:2017MiMic..23..155L. doi:10.1017/S1431927617000058. PMID 28228169. S2CID 6801783.
- ↑ Cheng, Yifan (2018-08-31). "Single-particle cryo-EM—How did it get here and where will it go". Science. 361 (6405): 876–880. Bibcode:2018Sci...361..876C. doi:10.1126/science.aat4346. ISSN 0036-8075. PMC 6460916. PMID 30166484.
- ↑ Sigworth, F.J. (1998). "A Maximum-Likelihood Approach to Single-Particle Image Refinement". Journal of Structural Biology. 122 (3): 328–339. doi:10.1006/jsbi.1998.4014. PMID 9774537.
- 1 2 Scheres, Sjors H.W. (January 2012). "A Bayesian View on Cryo-EM Structure Determination". Journal of Molecular Biology. 415 (2): 406–418. doi:10.1016/j.jmb.2011.11.010. PMC 3314964. PMID 22100448.
- 1 2 Nogales, Eva; Scheres, Sjors H.W. (May 2015). "Cryo-EM: A Unique Tool for the Visualization of Macromolecular Complexity". Molecular Cell. 58 (4): 677–689. doi:10.1016/j.molcel.2015.02.019. ISSN 1097-2765. PMC 4441764. PMID 26000851.
- ↑ Sigworth, Fred J. (2016-02-01). "Principles of cryo-EM single-particle image processing". Microscopy. 65 (1): 57–67. doi:10.1093/jmicro/dfv370. ISSN 2050-5698. PMC 4749045. PMID 26705325.
- ↑ Scheres, Sjors H. W. (2012-12-01). "RELION: Implementation of a Bayesian approach to cryo-EM structure determination". Journal of Structural Biology. 180 (3): 519–530. doi:10.1016/j.jsb.2012.09.006. ISSN 1047-8477. PMC 3690530. PMID 23000701.
- ↑ "RELION: Image-processing software for cryo-electron microscopy". GitHub. 3dem. 27 October 2023. Retrieved 27 October 2023.
- ↑ Bai, Xiao-chen; McMullan, Greg; Scheres, Sjors H.W (January 2015). "How cryo-EM is revolutionizing structural biology". Trends in Biochemical Sciences. 40 (1): 49–57. doi:10.1016/j.tibs.2014.10.005. ISSN 0968-0004. PMID 25544475. S2CID 19727349.
- ↑ Zivanov, Jasenko; Nakane, Takanori; Forsberg, Björn O; Kimanius, Dari; Hagen, Wim JH; Lindahl, Erik; Scheres, Sjors HW (2018-11-09). Egelman, Edward H; Kuriyan, John (eds.). "New tools for automated high-resolution cryo-EM structure determination in RELION-3". eLife. 7: e42166. doi:10.7554/eLife.42166. ISSN 2050-084X. PMC 6250425. PMID 30412051.
- ↑ Presentation on Cryoelectron Microscopy | PharmaXChange.info
- ↑ Shi D, Nannenga BL, Iadanza MG, Gonen T (November 2013). "Three-dimensional electron crystallography of protein microcrystals". eLife. 2: e01345. doi:10.7554/eLife.01345. PMC 3831942. PMID 24252878.
- ↑ Nannenga BL, Shi D, Leslie AG, Gonen T (September 2014). "High-resolution structure determination by continuous-rotation data collection in MicroED". Nature Methods. 11 (9): 927–930. doi:10.1038/nmeth.3043. PMC 4149488. PMID 25086503.
- ↑ Shi D, Nannenga BL, de la Cruz MJ, Liu J, Sawtelle S, Calero G, Reyes FE, Hattne J, Gonen T (May 2016). "The collection of MicroED data for macromolecular crystallography". Nature Protocols. 11 (5): 895–904. doi:10.1038/nprot.2016.046. PMC 5357465. PMID 27077331.
- ↑ de la Cruz MJ, Hattne J, Shi D, Seidler P, Rodriguez J, Reyes FE, Sawaya MR, Cascio D, Weiss SC, Kim SK, Hinck CS, Hinck AP, Calero G, Eisenberg D, Gonen T (February 2017). "Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED". Nature Methods. 14 (4): 399–402. doi:10.1038/nmeth.4178. PMC 5376236. PMID 28192420.
- ↑ Fu Z, Kaledhonkar S, Borg A, Sun M, Chen B, Grassucci RA, Ehrenberg M, Frank J (December 2016). "Key Intermediates in Ribosome Recycling Visualized by Time-Resolved Cryoelectron Microscopy". Structure. 24 (12): 2092–2101. doi:10.1016/j.str.2016.09.014. PMC 5143168. PMID 27818103.
- ↑ Feng X, Fu Z, Kaledhonkar S, Jia Y, Shah B, Jin A, Liu Z, Sun M, Chen B, Grassucci RA, Ren Y, Jiang H, Frank J, Lin Q (April 2017). "A Fast and Effective Microfluidic Spraying-Plunging Method for High-Resolution Single-Particle Cryo-EM". Structure. 25 (4): 663–670.e3. doi:10.1016/j.str.2017.02.005. PMC 5382802. PMID 28286002.
- ↑ Chen B, Kaledhonkar S, Sun M, Shen B, Lu Z, Barnard D, Lu TM, Gonzalez RL, Frank J (June 2015). "Structural dynamics of ribosome subunit association studied by mixing-spraying time-resolved cryogenic electron microscopy". Structure. 23 (6): 1097–105. doi:10.1016/j.str.2015.04.007. PMC 4456197. PMID 26004440.
Further reading
- Frank J (2006). Three-Dimensional Electron Microscopy of Macromolecular Assemblies. New York: Oxford University Press. ISBN 0-19-518218-9.
- van Heel M, Gowen B, Matadeen R, Orlova EV, Finn R, Pape T, Cohen D, Stark H, Schmidt R, Schatz M, Patwardhan A (November 2000). "Single-particle electron cryo-microscopy: towards atomic resolution". Quarterly Reviews of Biophysics. 33 (4): 307–69. doi:10.1017/s0033583500003644. PMID 11233408.
External links
| Library resources about Cryomicroscopy |
- "EM for Dummies". Retrieved June 9, 2006.
- The Fine Structure of a Frozen Virus – Sophisticated single-particle electron cryomicroscopy reveals unprecedented details in a virus's protein coat, Technology Review, March 19, 2008
- Getting Started in Cryo-EM – Online course from Caltech, Professor Grant Jensen
- EM Data Bank
- EMstats Trends and distributions of maps in EMDB, e.g. resolution trends
| Basics | |||||||
|---|---|---|---|---|---|---|---|
| Electron interaction with matter | |||||||
| Instrumentation | |||||||
| Microscopes |
| ||||||
| Techniques |
| ||||||
| Others |
| ||||||
| |||||||