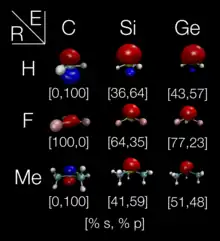
A trivalent group 14 radical (also known as a trivalent tetrel radical) is a molecule that contains a group 14 element (E = C, Si, Ge, Sn, Pb) with three bonds and a free radical, having the general formula of R3E•. Such compounds can be categorized into three different types, depending on the structure (or equivalently the orbital in which the unpaired electron resides) and the energetic barrier to inversion. A molecule that remains rigidly in a pyramidal structure has an electron in a sp3 orbital is denoted as Type A. A structure that is pyramidal, but flexible, is denoted as Type B. And a planar structure with an electron that typically would reside in a pure p orbital is denoted as Type C. The structure of such molecules has been determined by probing the nature of the orbital that the unpaired electron resides in using spectroscopy, as well as directly with X-ray methods.[3] Trivalent tetrel radicals tend to be synthesized from their tetravalent counterparts (i.e. R3EY where Y is a species that will dissociate).
Stability
While the trivalent triphenylmethyl radical, which was the first organic radical described, has been known for over 100 years,[4][5][6] characterization of transient, persistent, or stable radicals of heavier tetrel compounds have been only accessible in recent years (from the 1960s to the present). The most recent large advance has been the characterization of the first stable trivalent lead radical, as described in 2007.[3][7]
Such developments have only been made in recent years because these compounds tend to be highly reactive (with respect to reactions such as dimerization and radical chain reactions). There have been two main approaches for stabilization. Firstly electronic stabilization, the tetrel is connected to an electron-rich atom such as oxygen, nitrogen, or fluorine. Secondly steric stabilization, the tetrel is surrounded by bulky ligands (such as -Y(SiMe3)2 (Y = N, CH), -Si(SiMe3)2Et (-Ebt), or -Si(SiMe3)3 (-Hyp)). It has become convention to describe a radical that can persist long enough for spectroscopic or chemical analysis as persistent and a radical that can persist indefinitely as stable.[7] Trivalent tetrels can also synthesized in a cyclic structure (e.g. Ar3Ge3•). This class of molecules tends to be slightly more stable than the acyclic analogues as there is a stabilization through the delocalization of the unpaired electrons within the π-system.[8]
Synthetic methodology
Trivalent radicals can be prepared from the tetrel hydride (for arbitrary radical species Z).[9]
- Z• + R3EH → ZH + R3E•
They can also be formed by oxidation of the salt (typically with GeCl2•dioxane in Et2O).[10]
- R3ENa + ECl2•dioxane → R3E•
They can be formed via photolysis.[10]
- R3EH + hν → R3E•
- R3Si-SiR3 + hν → 2 R3E•
Or they can be formed via thermal disproportionation (thermolysis) of the related dimeric species.[11]
- R2EER2 → ER3• + ER•
These can also be formed via gamma-irradiation of an ER4 complex.[8][12]
- R4E + hν → R3E• + R•
As well as by a reduction pathway.[8]
- R3ECl + Na → R3E• + NaCl
Spectroscopy and characterization
Electronic spectroscopy
Information about the structure of these trivalent tetrels has been determined by mainly EPR spectroscopy and X-ray crystallography, however the geometry of transient small molecules has been determined via resonance-enhanced multiphoton ionization, transient UV absorption spectroscopy, and microwave spectroscopy by determining vibrational and rotational resonance frequencies.[8]
ESR/EPR spectroscopy
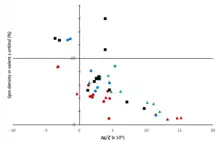
Electron paramagnetic resonance has been paramount for the study of trivalent tetrels as the hyperfine coupling to the tetrel reveals the orbital in which the unpaired electron resides, and the orbital composition directly correlates to the structure of the molecule. The isotropic component of the hyperfine coupling to the central tetrel scales proportionally with the spin density in the valence s orbital on that atom (see the Figure on the right). By comparing this isotropic hyperfine coupling constant to the theoretical hyperfine splitting of an electron in a pure valence s orbital,[19] one can calculate the percent of the unpaired spin density in the valence s orbital. Similarly, the ratio of the anisotropic hyperfine coupling constant to the anisotropic hyperfine coupling of a single electron in a pure atomic p orbital reveals the percent of spin occupation in a valence p orbital. However, measurement of the anisotropic component of the hyperfine tensor are more difficult and not as frequent in literature.
The percent of spin occupation in the valence s orbital can be used to directly probe the structure of these molecules. If the spin occupation 100% in a p orbital, then the molecule will have a Type C planar structure. However, if there is 25% s orbital and 75% p orbital occupation, then the molecule will have a pyramidal Type A structure. Any intermediate value is possible and would correspond to a Type B structure. Values of greater than 25% s orbital contribution can also be found upon coordination of a tetrel to electronegative ligands (-OR, -F, -NR2, -Cl).[13]
There is also a correlation between the g-shift (∆g = gmeas - ge) and the geometry for series of compounds with ligands of similar electronegativities. More electronegative ligands correspond to more tetrahedral geometries. Lower g values correlate more with pyramidal structures, while higher g values correlate with planar structures.[11][20] It has also been demonstrated using tris(trialkylsilyl)silyl radicals that the more bulky the ligands are, the more a planar structure will be favored, and the lower the hyperfine coupling constant will be.[21]
Theoretical calculations
It has been shown that there are two main factors that dictate whether a complex will be a Type A, B, or C structure. The lighter the tetrel, the more it will have a tendency to remain planar. This has been ascribed due to the pseudo Jahn–Teller effect, as the E-R anti-bonding orbitals (of R3E•) can more significantly mix with the non-bonding SOMO (singly occupied molecular orbital) due to a more electropositive and diffuse central atom. The barrier for inversion has been calculated at the NL-SCF/TZ2P level to be increasing for EH3• C, Si, Ge, Sn at 0.0, 3.7, 3.8, 7.0 kcal/mol (the barrier for inversion of methyl radical is zero as it is most stable in a planar Type C structure).[22]
References
- ↑ Bochevarov, Art D; Harder, Edward; Hughes, Thomas F; Greenwood, Jeremy; Braden, Dale A; Phillipp, Dean M; Rinaldo, David; Halls, Mathew D; Zhang, Jing; Friesner, Richard A (2013). "Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences". International Journal of Quantum Chemistry. 113 (18): 2110–2142. doi:10.1002/qua.24481.
- ↑ Ohta, K; Nakatsuji, H; maeda, I; Yonezawa, T (1982). "Ab initio calculation of geometries and hfs constants of CH3, SiH3 and GeH3 radicals". Chemical Physics. 67 (1): 49–58. Bibcode:1982CP.....67...49O. doi:10.1016/0301-0104(82)88057-9.
- 1 2 Lee, Vladimir Ya.; Sekiguchi, Akira. (2005-04-07). "Si‐, Ge‐, and Sn‐Centered Free Radicals: From Phantom Species toGrams‐Order‐Scale Materials". European Journal of Inorganic Chemistry. 2005 (7): 1209–1222. doi:10.1002/ejic.200400999.
- ↑ Gomberg, M. (1900). "An instance of trivalent carbon: triphenylmethyl". Journal of the American Chemical Society. 22 (11): 757–771. doi:10.1021/ja02049a006.
- ↑ Gomberg, M. (1901). "On trivalent carbon". Journal of the American Chemical Society. 23 (7): 496–502. Bibcode:1901Sci....13..116N. doi:10.1021/ja02033a015.
- ↑ Gomberg, M. (1902). "On trivalent carbon". Journal of the American Chemical Society. 24 (7): 597–628. doi:10.1021/ja02021a001.
- 1 2 Förster, Christoph; Klinkhammer, Karl W.; Tumanskii, Boris; Krüger, Hans-Jörg; Kelm, Harald (2007-01-29). "Stable Mononuclear Lead(III) Compound: A Lead‐Centered Radical". Angewandte Chemie International Edition. 46 (7): 1156–1159. doi:10.1002/anie.200603323. PMID 17171746.
- 1 2 3 4 Lee, Vladimir Ya.; Sekiguchi, Akira (2006-09-12). "2". Silicon‐, Germanium‐, and Tin‐Centered Cations, Radicals, and Anions. pp. 47–120. doi:10.1002/9780470120828.ch2. ISBN 9780470120828.
{{cite encyclopedia}}:|journal=ignored (help) - 1 2 Chatgilialoglu, Chryssostomos (1995-06-01). "Structural and Chemical Properties of Silyl Radicals". Chemical Reviews. 95 (5): 1229–1251. doi:10.1021/cr00037a005.
- 1 2 Chatgilialoglu, C.; Schiesser, C.H. (2009-12-15). "Silyl Radicals". PATai's Chemistry of Functional Groups. doi:10.1002/9780470682531.pat0238. ISBN 9780470682531.
- 1 2 Hudson, Andrew; Lapper, Michael F.; Lednor, Peter W. (1976). "Subvalent Group 4B metal alkyls and amides. Part 4. An electron spin resonance study of some long-lived photochemically synthesised trisubstituted silyl, germyl, and stannyl radicals". J. Chem. Soc., Dalton Trans. 22 (22): 2369. doi:10.1039/DT9760002369.
- ↑ Jackel, George S.; Gordy, Walter (1968-12-10). "Electron Spin Resonance of Free Radicals Formed from Group-IV and Group-V Hydrides in Inert Matrices at Low Temperature". Phys. Rev. 176 (443): 443–452. Bibcode:1968PhRv..176..443J. doi:10.1103/PhysRev.176.443.
- 1 2 Iley, Jim (1995-08-15). "5". CESR of Organogermanium, Organotin and Organolead Radicals. The Chemistry of Organic Germanium, Tin and Lead Compounds, Volume 1. The Chemistry of Functional Groups. Vol. 1. pp. 267–289. doi:10.1002/0470857242.ch5. ISBN 0471942073.
- ↑ Becker, Marco; FöRster, Christoph; Franzen, Christian; Hartrath, Johannes; Kirsten, Enzio; Knuth, Jörn; Klinkhammer, Karl W.; Sharma, Ajay; Hinderberger, Dariush (2008-09-30). Persistent Radicals of Trivalent Tin and Lead. Vol. 47. pp. 9965–9978. doi:10.1021/ic801198p. PMID 18823115.
{{cite encyclopedia}}:|journal=ignored (help) - ↑ Gouterman, Martin; Schwarz, Frederick; Smith, Paul (1973). "Porphyrins. XXVII. Spin‐orbit coupling and luminescence of Group IV complexes". J. Chem. Phys. 59 (2): 676–690. Bibcode:1973JChPh..59..676G. doi:10.1063/1.1680075.
- ↑ Moore, Charlotte (1949). Atomic Energy Levels. U.S. Natl. Bur. Stand.
- ↑ Moore, Charlotte (1952). Atomic Energy Levels. U.S. Natl. Bur. Stand.
- ↑ Moore, Charlotte (1958). Atomic Energy Levels. U.S. Natl. Bur. Stand.
- ↑ Morton, JR; Preston, KF (1978). "Atomic parameters for paramagnetic resonance data". Journal of Magnetic Resonance. 30 (3): 577–582. Bibcode:1978JMagR..30..577M. doi:10.1016/0022-2364(78)90284-6.
- ↑ Gynane, Michael; Harris, David; Lappert, Michael; Power, Philip; Riviere, Pierre; Rivere-Baudet, Monique (1977). "Subvalent Group 4B metal alkyls and amides. Part 5. The synthesis and physical properties of thermally stable amides of germanium(II), tin(II), and lead(II)". Journal of the Chemical Society, Dalton Transactions (20): 2004–2009. doi:10.1039/DT9770002004.
- ↑ Kyushin, Soichiro; Sakurai, Haruaki; Betsuyaku, Takashi; Matsumoto, Hideyuki (1997-12-09). "Highly Stable Silyl Radicals (EtnMe3−nSi)3Si• (n = 1−3)". Organometallics. 16 (25): 5386–5388. doi:10.1021/om970607x.
- ↑ Bickelhaupt, F. Matthias; Ziegler, Tom; von Ragué Schleyer, Paul (1996-03-05). "CH3• Is Planar Due to H−H Steric Repulsion. Theoretical Study of MH3• and MH3Cl (M = C, Si, Ge, Sn)". Organometallics. 15 (5): 1477–1487. doi:10.1021/om950560k.