Whole genome bisulfite sequencing is a next-generation sequencing technology used to determine the DNA methylation status of single cytosines by treating the DNA with sodium bisulfite before high-throughput DNA sequencing. The DNA methylation status at various genes can reveal information regarding gene regulation and transcriptional activities.[1] This technique was developed in 2009 along with reduced representation bisulfite sequencing after bisulfite sequencing became the gold standard for DNA methylation analysis.[2][3]
Whole genome bisulfite sequencing measures single-cytosine methylation levels genome-wide and directly estimates the ratio of molecules methylated rather than enrichment levels. Currently, this technique has recognized and tested approximately 95% of all cytosines in known genomes.[4] With the improvement of library preparation methods and next-generation sequencing technology over the past decade, whole genome bisulfite sequencing has become an increasingly widespread and informative method for analyzing DNA methylation in epigenomic-wide studies.[5]
History
Prior to the development of whole genome bisulfite sequencing, genome methylation analysis relied heavily on early non-specific and differential methods such as paper chromatography, high-performance liquid chromatography, and thin-layer chromatography to analyze methylation profiles.[6] These methods were limited by the inability to amplify methylated DNA via polymerase chain reaction in vitro due to loss of methylation status.[6] As a result, much of these early methods relied on detecting and analyzing naturally-manifested methylated cytosines in vivo rather than chemically methylated cytosines.
In 1970, a breakthrough occurred when it was discovered that treating DNA with sodium bisulfite deaminated cytosine residues into uracil.[6] In the following decade, this discovery led to the revelation that unmethylated cytosine reacted much faster to sodium bisulfite treatment than did 5-methylcytosine. This difference in reaction rates created the possibility of identifying chemical changes in DNA as an easily detectable genetic marker.[6] Whole genome bisulfite sequencing was derived as a combination of this bisulfite treatment and next-generation sequencing technology, such as shotgun sequencing.
The whole genome sequencing technique was first applied to the DNA methylation mapping at single nucleotide resolution to Arabidopsis thaliana in 2008, and shortly after in 2009, the first single-base-resolution DNA methylation map of the entire human genome was created using whole genome bisulfite sequencing.[7][5] Since its development, many various protocols of whole genome bisulfite sequencing have been developed aiming to improve the efficiency and efficacy of its single-base mapping. As the costs of next-generation sequencing have decreased, whole genome bisulfite sequencing has become more widely used in clinical and experimental research.[3] Currently, multiple public datasets of genomic data have been established, and this technique has recognized and tested approximately 95% of all cytosines in known genomes.[4]
Method
The following steps are derived from one potential workflow of conventional whole genome bisulfite sequencing: target DNA extraction, bisulfite conversion, library amplification, and bioinformatics analysis.[8] However, various sequencing systems and analysis tools often adapt the technical parameters and order of the following step processes in order to optimize assay coverage and efficacy.[3]
DNA extraction
Library preparation protocols undergo DNA fragmentation, end repair, dA-tailing, and adapter ligation prior to bisulfite treatment and library amplification. Standard fragmentation under high-throughput technology such as Illumina Genome Analyser and Solexa requires nebulization to generate fragments that range from 0-1200 base pairs.[9] After fragmentation, end repair enzymes and complementary adapters are then applied to the DNA in an end-prep polymerase chain reaction and adapter ligation reaction, respectively. Size selection occurs before the DNA is treated with sodium bisulfite.
Conventional methods of eukaryotic DNA preparation during sequencing use a wide variety of DNA input amount, varying from as little as 10 ng for novel NGS library alternatives, such as the tagmentation approach, to as much as 500-1000 ng of DNA as sample input.[10]
Bisulfite conversion
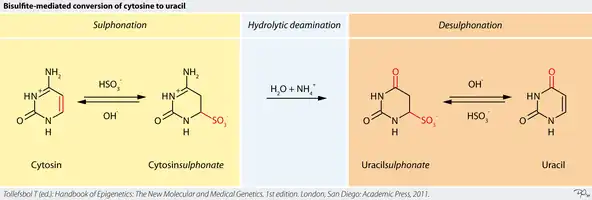
The adapter-ligated DNA sample is treated with sodium bisulfite, a chemical compound that converts unmethylated cytosines into uracil, at low pH and high temperatures.[11][12] The chemical reaction is depicted in Figure 1, where sulfonation occurs at the carbon-6 position of cytosine to produce the intermediate cytosine sulfonate.[13] This intermediate then undergoes irreversible hydrolytic deamination to create uracil sulfonate. Under alkaline conditions, uracil sulfonate desulfonates to generate uracil.[13]
This enables methylation detection by distinguishing the methylated cytosines (5-methylcytosine), which resist bisulfite treatment, from uracil. During amplification by polymerase chain reaction, the uracils are converted into thymines.[3] Methylated cytosines are then recognized as cytosines. Their locations are then identified by comparison of the bisulfite-treated and original DNA sequence.
Following bisulfite treatment, purification of the sample is required to remove unwanted products including bisulfite salts.[13]
Library amplification
In order to amplify the epigenome library, bisulfite-treated DNA is primed to generate DNA with a specific tagging sequence. The 3' end of this sequence is then tagged again, creating DNA fragments with markers on either end. These fragments are amplified in a final polymerase chain reaction reaction, after which the library is prepped for sequencing-by-synthesis.[8] This is demonstrated in Figure 2, in which high-throughput sequencing system developed by biotechnology company, Illumina, perform comprehensive assays based on sequencing-by-synthesis of base pairs.[8]
Bioinformatics analysis
Following library amplification, a series of analyses can be performed on the expanded library to determine various methylation characteristics or map a genome-wide methylation profile.[8]
One such study aligns the new reads against the reference genome in order to directly compare locations of methylated cytosines and C-T mismatches. This requires software such as SOAP for side-by-side comparison of the genomes.[8] Another potential sequencing analysis is methylated cytosine calling, which computes methylated cytosine ratios by mapping probabilities based on read quality. This helps determine methylated cytosine locations across the genome.[8] Finally, global trends of methylome can be analyzed by calculating the distribution ratios of CG, CHGG, and CHH in methylated cytosines across the genome.[8] These ratios can reflect features of whole genome methylation maps of certain species.

Applications
Due to its ability to screen methylation status at singe-nucleotide resolution across a given genome, whole genome bisulfite sequencing has become increasingly promising in aiding fundamental epigenomics research, novel hypotheses on DNA methylation, and investigations of future large-scale epidemiological studies.[3][5] This whole genome approach is also capable of sensitive cytosine-methylation detection under specific sequences across an entire genome, which increases its potential to identify specific DNA methylation sites and their relation to certain gene expressions.[6]
DNA Methylation
The whole genome bisulfite sequencing technique is capable of sensitive cytosine-methylation detection under specific sequences across an entire genome, which increases its potential to identify specific DNA methylation sites and their relation to certain gene expressions.[6] The use of whole genome bisulfite sequencing to create the first human DNA methylome in 2009 also helped identify a significant ratio of non-CG methylation.[6] As a result, multiple single-base resolution methylomes of the human genome continue to be produced in order to identify the role of intragenic DNA methylation in gene expression and regulation. Future studies aim to use whole genome bisulfite sequencing in order to investigate the role DNA methylation has in multifarious cellular processes such as cellular differentiation, embryogenesis, X-inactivation, genomic imprinting, and tumorigenesis.[4] Single-nucleotide maps have already been sequenced for two human cell lines, H1 human embryonic stem cells and IMR90 fetal lung fibroblasts, in order to study patterns of non-CG methylation in human cells.[4]
Developmental biology
Whole genome bisulfite sequencing has also been applied to developmental biology studies in which non-CG methylation was discovered prevalent in pluripotent stem cells and oocytes. This technique helped researchers discover that non-CG methylation accumulated during oocyte growth and covered over half of all methylation in mouse germinal vesicle oocytes.[14] Similarly, in plants, whole genome bisulfite sequencing was used to examine CG, CHH, and CHG methylation. It was then discovered that the plant germline conserved CG and CHG methylation while mammals lost CHH methylation in microspores and sperm cells.[14]
Other fields
The unlimited resources provided by the approach of an entire genome have spurred many novel hypotheses on how whole genome bisulfite sequencing could be used in other various fields including disease diagnosis and forensic science. Studies have shown that whole genome bisulfite sequencing could detect abnormal methylation, or more specifically hyper-methylated suppressor genes, that are often seen in cancers including leukemia.[14] Additionally, whole genome bisulfite sequencing has been applied to blood spot samples in forensic investigations to generate high-quality DNA methylation analyses on dried stains.[14]
Limitations
Technical concerns
The widespread use of whole genome bisulfite sequencing has been primarily limited by its excessive cost, complex data output, and minimal required coverage. Due to the high amount and subsequent cost of DNA input, many studies using whole genome bisulfite sequencing assays occur with few or no biological replicates.[15] For human samples, the US National Institutes of Health (NIH) Roadmap Epigenomics Project recommends a minimum of 30x coverage sequencing to achieve accurate results and approximately 80 million aligned, high quality reads.[16] Consequently, large-scale studies for genomic-wide methylation profiling remain less cost-effective, often requiring multiple re-sequences of the entire genome multiple times for every experiment.[17] Current studies are being conducted to reduce the conventional minimum coverage requirements while maintaining mapping accuracy.
Finally, the technique is also limited the complexity of data and lack of sufficiently advanced analytical tools for downstream computational requirements.[2] The current bioinformatics requirements for accurate data interpretation are ahead of existing technology, which stalls the accessibility of sequencing results to the general public.
Biases and over-representation of DNA methylation
Additionally, there are biological limitations concerning various steps in the standard protocol, particularly in the library preparation method. One of the biggest concerns is the potential of bias in the base composition of sequences and over-representation of methylated DNA data following bioinformatics analyses.[9] Bias can arise from multiple unintended effects of bisulfite conversion including DNA degradation. This degradation can cause uneven sequence coverage by misrepresenting genomic sequences and overestimating 5-methylcytosine values.[3] Additionally, the bisulfite conversion process only distinguishes unmethylated cytosine from 5-methylcytosine. As a result, specificity between 5-methylcytosine and 5-hydroxymethylcytosine is limited.[3] Another potential source of bias rises from polymerase chain reaction amplification of the library, which affects sequences with highly skewed base compositions due to high rates of polymerase sequence errors in high AT-content, bisulfite-converted DNA.[3]
See also
References
- 1 2 Kawakatsu, Taiji (2020), Vaschetto, Luis M. (ed.), "Whole-Genome Bisulfite Sequencing and Epigenetic Variation in Cereal Methylomes", Cereal Genomics: Methods and Protocols, Methods in Molecular Biology, New York, NY: Springer US, vol. 2072, pp. 119–128, doi:10.1007/978-1-4939-9865-4_10, ISBN 978-1-4939-9865-4, PMID 31541442, S2CID 202711452, retrieved 2021-11-14
- 1 2 Stirzaker, Clare; Taberlay, Phillippa C.; Statham, Aaron L.; Clark, Susan J. (2014-02-01). "Mining cancer methylomes: prospects and challenges". Trends in Genetics. 30 (2): 75–84. doi:10.1016/j.tig.2013.11.004. ISSN 0168-9525. PMID 24368016.
- 1 2 3 4 5 6 7 8 Olova, Nelly; Krueger, Felix; Andrews, Simon; Oxley, David; Berrens, Rebecca V.; Branco, Miguel R.; Reik, Wolf (2018-03-15). "Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data". Genome Biology. 19 (1): 33. doi:10.1186/s13059-018-1408-2. ISSN 1474-760X. PMC 5856372. PMID 29544553.
- 1 2 3 4 Lister, Ryan; Pelizzola, Mattia; Dowen, Robert H.; Hawkins, R. David; Hon, Gary; Tonti-Filippini, Julian; Nery, Joseph R.; Lee, Leonard; Ye, Zhen; Ngo, Que-Minh; Edsall, Lee (2009-10-14). "Human DNA methylomes at base resolution show widespread epigenomic differences". Nature. 462 (7271): 315–322. Bibcode:2009Natur.462..315L. doi:10.1038/nature08514. ISSN 1476-4687. PMC 2857523. PMID 19829295.
- 1 2 3 Zhou, Li; Ng, Hong Kiat; Drautz-Moses, Daniela I.; Schuster, Stephan C.; Beck, Stephan; Kim, Changhoon; Chambers, John Campbell; Loh, Marie (2019-07-17). "Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing". Scientific Reports. 9 (1): 10383. Bibcode:2019NatSR...910383Z. doi:10.1038/s41598-019-46875-5. ISSN 2045-2322. PMC 6637168. PMID 31316107.
- 1 2 3 4 5 6 7 Parle-Mcdermott, Anne; Harrison, Alan (2011). "DNA Methylation: A Timeline of Methods and Applications". Frontiers in Genetics. 2: 74. doi:10.3389/fgene.2011.00074. ISSN 1664-8021. PMC 3268627. PMID 22303369.
- ↑ Cokus, Shawn J.; Feng, Suhua; Zhang, Xiaoyu; Chen, Zugen; Merriman, Barry; Haudenschild, Christian D.; Pradhan, Sriharsa; Nelson, Stanley F.; Pellegrini, Matteo; Jacobsen, Steven E. (2008-02-17). "Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning". Nature. 452 (7184): 215–219. Bibcode:2008Natur.452..215C. doi:10.1038/nature06745. ISSN 1476-4687. PMC 2377394. PMID 18278030.
- 1 2 3 4 5 6 7 8 "Principles and Workflow of Whole Genome Bisulfite Sequencing – CD Genomics". www.cd-genomics.com. Retrieved 2021-11-03.
- 1 2 Quail, Michael A.; Kozarewa, Iwanka; Smith, Frances; Scally, Aylwyn; Stephens, Philip J.; Durbin, Richard; Swerdlow, Harold; Turner, Daniel J. (2008-11-25). "A large genome center's improvements to the Illumina sequencing system". Nature Methods. 5 (12): 1005–1010. doi:10.1038/nmeth.1270. ISSN 1548-7105. PMC 2610436. PMID 19034268.
- ↑ Wang, Qi; Gu, Lei; Adey, Andrew; Radlwimmer, Bernhard; Wang, Wei; Hovestadt, Volker; Bähr, Marion; Wolf, Stephan; Shendure, Jay; Eils, Roland; Plass, Christoph (2013-09-26). "Tagmentation-based whole-genome bisulfite sequencing". Nature Protocols. 8 (10): 2022–2032. doi:10.1038/nprot.2013.118. ISSN 1750-2799. PMID 24071908. S2CID 12706151.
- ↑ Frommer, M; McDonald, L E; Millar, D S; Collis, C M; Watt, F; Grigg, G W; Molloy, P L; Paul, C L (1992-03-01). "A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands". Proceedings of the National Academy of Sciences of the United States of America. 89 (5): 1827–1831. Bibcode:1992PNAS...89.1827F. doi:10.1073/pnas.89.5.1827. ISSN 0027-8424. PMC 48546. PMID 1542678.
- ↑ Clark, S J; Harrison, J; Paul, C L; Frommer, M (1994-08-11). "High sensitivity mapping of methylated cytosines". Nucleic Acids Research. 22 (15): 2990–2997. doi:10.1093/nar/22.15.2990. ISSN 0305-1048. PMC 310266. PMID 8065911.
- 1 2 3 Kristensen, Lasse Sommer; Hansen, Lise (2009-07-01). "PCR-Based Methods for Detecting Single-Locus DNA Methylation Biomarkers in Cancer Diagnostics, Prognostics, and Response to Treatment". Clinical Chemistry. 55 (8): 1471–83. doi:10.1373/clinchem.2008.121962. PMID 19520761.
- 1 2 3 4 "Applications of Whole Genome Bisulfite Sequencing (WGBS)". News-Medical.net. 2018-10-31. Retrieved 2021-11-17.
- ↑ Wu, Hao; Xu, Tianlei; Feng, Hao; Chen, Li; Li, Ben; Yao, Bing; Qin, Zhaohui; Jin, Peng; Conneely, Karen N. (2015-12-02). "Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates". Nucleic Acids Research. 43 (21): e141. doi:10.1093/nar/gkv715. ISSN 0305-1048. PMC 4666378. PMID 26184873.
- ↑ Ziller, Michael J.; Hansen, Kasper D.; Meissner, Alexander; Aryee, Martin J. (2014-11-02). "Coverage recommendations for methylation analysis by whole-genome bisulfite sequencing". Nature Methods. 12 (3): 230–232. doi:10.1038/nmeth.3152. ISSN 1548-7105. PMC 4344394. PMID 25362363.
- ↑ Stevens, Michael; Cheng, Jeffrey B.; Li, Daofeng; Xie, Mingchao; Hong, Chibo; Maire, Cécile L.; Ligon, Keith L.; Hirst, Martin; Marra, Marco A.; Costello, Joseph F.; Wang, Ting (2013-09-23). "Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods". Genome Research. 23 (9): 1541–1553. doi:10.1101/gr.152231.112. ISSN 1088-9051. PMC 3759729. PMID 23804401.