




ATP-dependent metalloprotease YME1L1 is an enzyme that in humans is encoded by the YME1L1 gene.[5] YME1L1 belongs to the AAA family of ATPases and mainly functions in the maintenance of mitochondrial morphology. Mutations in this gene would cause infantile-onset mitochondriopathy.[6]
Structure
Gene
The YME1L1 gene is located at chromosome 10p14, consisting of 20 exons. Two transcript variants encoding different isoforms have been found for this gene.
Protein
YME1L1 consists of 716 amino acids and is highly similar to all mitochondrial AAA proteases and in particular to yeast Yme1p. Three different domains are identified via sequence analysis, including an AAA consensus sequence between amino acids 317 and 502, an ATP/GTP binding motif, and a HEXXH motif typical of a zinc-dependent binding domain.[7]
Function
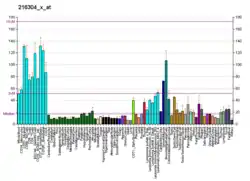
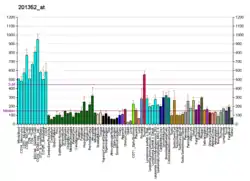
YME1L1 is embedded in the inner mitochondrial membrane and is more abundant in tissues with a high content of mitochondria such as human adult heart, skeletal muscle, and pancreas RNA.[7][6] YME1L1 is a member of the AAA family of ATPases and has an important role for the maintenance of mitochondrial morphology.[6] Its mature form assembles into a homo-oligomeric complex within the inner mitochondrial membrane (IM).[8] It degrades both intermembrane space and IM proteins, including lipid transfer proteins, components of protein translocases of the IM, and the dynamin-like GTPase optic atrophy 1 (OPA1) [9][10][11] Loss of YME1L1 accelerates OMA1-dependent long-form OPA1 cleavage, resulting in short-form OPA1 accumulation, increased mitochondrial fission, and mitochondrial network fragmentation.[12] It's also reported that YME1L1 controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation.[13]
Clinical significance
A homozygous mutation in the YME1L1 gene would cause infantile-onset mitochondriopathy, with severe intellectual disability, muscular impairments, and optic nerve atrophy. The missense mutation affects the MPP processing site and impairs YME1L1 maturation, leading to its rapid degradation, and also leads to a proliferation defect, abnormal OPA1 processing and mitochondrial fragmentation.[6]
Interactions
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000136758 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000026775 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Entrez Gene: YME1L1 YME1-like 1 (S. cerevisiae)".
- 1 2 3 4 Hartmann B, Wai T, Hu H, MacVicar T, Musante L, Fischer-Zirnsak B, Stenzel W, Gräf R, van den Heuvel L, Ropers HH, Wienker TF, Hübner C, Langer T, Kaindl AM (6 August 2016). "Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation". eLife. 5. doi:10.7554/eLife.16078. PMC 4991934. PMID 27495975.
- 1 2 Coppola M, Pizzigoni A, Banfi S, Bassi MT, Casari G, Incerti B (May 2000). "Identification and characterization of YME1L1, a novel paraplegin-related gene". Genomics. 66 (1): 48–54. doi:10.1006/geno.2000.6136. PMID 10843804.
- ↑ Van Dyck L, Langer T (November 1999). "ATP-dependent proteases controlling mitochondrial function in the yeast Saccharomyces cerevisiae". Cellular and Molecular Life Sciences. 56 (9–10): 825–42. doi:10.1007/s000180050029. PMID 11212342. S2CID 5825456.
- ↑ Potting C, Tatsuta T, König T, Haag M, Wai T, Aaltonen MJ, Langer T (August 2013). "TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid". Cell Metabolism. 18 (2): 287–95. doi:10.1016/j.cmet.2013.07.008. PMID 23931759.
- 1 2 Rainbolt TK, Saunders JM, Wiseman RL (January 2015). "YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress". EMBO Reports. 16 (1): 97–106. doi:10.15252/embr.201438976. PMC 4304733. PMID 25433032.
- ↑ Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (March 2014). "The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission". The Journal of Cell Biology. 204 (6): 919–29. doi:10.1083/jcb.201308006. PMC 3998800. PMID 24616225.
- ↑ Wai T, García-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Rupérez FJ, Barbas C, Ibañez B, Langer T (December 2015). "Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice". Science. 350 (6265): aad0116. doi:10.1126/science.aad0116. PMID 26785494. S2CID 34322149.
- ↑ Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, Houstek J, Zeman J (March 2012). "YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation". Molecular Biology of the Cell. 23 (6): 1010–23. doi:10.1091/mbc.E11-08-0674. PMC 3302729. PMID 22262461.
- ↑ MacVicar T, Langer T (June 2016). "OPA1 processing in cell death and disease - the long and short of it". Journal of Cell Science. 129 (12): 2297–306. doi:10.1242/jcs.159186. PMID 27189080.
Further reading
- Shah ZH, Hakkaart GA, Arku B, de Jong L, van der Spek H, Grivell LA, Jacobs HT (August 2000). "The human homologue of the yeast mitochondrial AAA metalloprotease Yme1p complements a yeast yme1 disruptant". FEBS Letters. 478 (3): 267–70. doi:10.1016/S0014-5793(00)01859-7. PMID 10930580. S2CID 42014862.
- Zhang QH, Ye M, Wu XY, Ren SX, Zhao M, Zhao CJ, Fu G, Shen Y, Fan HY, Lu G, Zhong M, Xu XR, Han ZG, Zhang JW, Tao J, Huang QH, Zhou J, Hu GX, Gu J, Chen SJ, Chen Z (October 2000). "Cloning and functional analysis of cDNAs with open reading frames for 300 previously undefined genes expressed in CD34+ hematopoietic stem/progenitor cells". Genome Research. 10 (10): 1546–60. doi:10.1101/gr.140200. PMC 310934. PMID 11042152.
- Pellegrini L, Passer BJ, Canelles M, Lefterov I, Ganjei JK, Fowlkes BJ, Koonin EV, D'Adamio L (April 2001). "PAMP and PARL, two novel putative metalloproteases interacting with the COOH-terminus of Presenilin-1 and -2". Journal of Alzheimer's Disease. 3 (2): 181–190. doi:10.3233/jad-2001-3203. PMID 12214059.