硅醚
硅醚是含有硅原子与烷氧基以共价键键合的一类化合物。其通式为:R1R2R3Si−O−R4,其中R4为烷基取代基或芳基取代基。硅醚在有机合成当中常用于醇的保护基。R1R2R3基团可以是不同的烃基基团,因此可组合成多样的硅醚。硅醚化合物在保护基化学当中有非常广泛的应用,常用的硅醚有:三甲基硅基 (TMS),叔丁基二苯基硅基(TBDPS),叔丁基二甲基硅基(TBS/TBDMS)和三异丙基硅基(TIPS)。

形成
制备硅醚的方法很多,但大多可遵循两种策略:醇与氯硅烷在胺作碱的条件下室温反应,或醇与三氟甲磺酸硅酯在大位阻碱的条件下低温反应。三氟甲磺酸硅酯比起相应的氯硅烷活性更强,因此常使用三氟甲磺酸硅酯保护位阻较大的羟基。另一种非常可靠且快捷的方法是Corey法,即咪唑作碱将醇与氯硅基在二甲基甲醯胺(DMF)溶剂中高浓度反应[1]。
如果使用二氯甲烷代替DMF作为溶剂,反应速率会相对降低但更易于纯化产物。使用三氟甲磺酸硅酯取代时,常用的大位阻碱是2,6-二甲基吡啶[2],伯醇的硅醚保护通常在一小时内可反应完全,而大位阻的醇反应完全则常需要几天时间。
当使用氯硅烷进行取代时对于反应条件通常不苛刻,只需避免在反应中引入大量的水。过量的氯硅烷可加速反应进程但并非必须。若使用了大量的硅代试剂,后处理则需要使用色谱法进行分离纯化,以去除过量的硅醇和硅氧烷化合物。三氟甲磺酸硅酯是一类水敏感化合物,因此必须在惰性气体保护下进行反应,后处理通常需要使用酸性水溶液如饱和氯化铵溶液进行淬灭。这种淬灭法能够不破坏硅试剂而质子化胺碱,让其移除反应体系。最后进行萃取操作并进行柱层析纯化。
三氟甲磺酸硅酯活性很强,甚至可以转化酮成为硅基烯醇醚化合物。
硅醚保护基的脱除
当不需要硅醚保护时,可使用酸或者氟试剂如四正丁基氟化铵进行保护基的脱除[3]。通常硅醚取代基体积越大抵御水解能力越强,但引入这种取代基相对越困难[4]。
在酸性体系中,硅醚的相对稳定性如下(参见上述参考文献):
- TMS (1) < TES (64) < TBS (20 000) < TIPS (700 000) < TBDPS (5 000 000)
在碱性体系中,硅醚的相对稳定性如下(参见上述参考文献):
- TMS (1) < TES (10-100) < TBS~TBDPS (20 000) < TIPS (100 000)
对称二醇的单保护
对称二醇的单硅醚化理论上是可行的,但实际操作中存在着诸多问题。报道的单硅取代反应举例如下:[5]
单取代反应的主要问题在于这类反应难于重复。如果反应仅是热力学控制的,且双负离子的活性与单负离子相近,那么相应得到的混合产物(双取代:单取代:非取代原料)的比例应为:1:2:1。而如果使用四氢呋喃作溶剂,反应会受到溶剂对双负离子的脱质子化的影响,它包括了:动力学因素影响以及溶剂对于单负离子的溶解度较低的影响。反应初始加入TBSCl时,溶液中只会溶解少量的单负离子而其余的则无法溶解形成悬浮液。仅仅这部分溶解的单负离子参与反应,而使得反应平衡向形成更多单负离子的方向移动,最后允许反应得到较高收率的单取代-TBS的产物。有使用正丁基锂作碱而取得更好收率的例子[6]:
第三种方法是使用DMF与DIPEA的混合物体系进行反应[7]。
另一种策略是使用过量的二醇进行反应,如使用4.0当量迫使反应朝着单取代的方向进行。
选择性脱保护
选择性的脱硅基保护在许多情况下是可行的,例如:[8]。
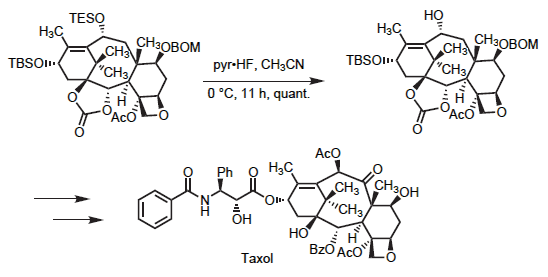
硅醚之间的区别主要基于位阻及电性的不同。总的来讲,酸性条件脱除位阻较小的硅基团相对更快,而位阻较大的硅基团则因为氧原子上具有很大的空间保护而不易脱除。基于氟的脱保护对于缺电子的硅基团则快于富电子的硅基团。有一些证据表明,硅基脱保护过程是通过高价硅物种进行的。
虽然选择性脱去硅醚保护可通过诸多条件,但以下列举的一些条件相对较可靠。若两种硅取代基存在着较大的位阻区别则选择性脱除相对更易成功,如:一级TBS与二级TBS或一级TES与一级TBS,或电性有区别,如:一级TBDPS与一级TBS。虽然如此,但有些时候反复的优化条件是不可避免的,有时甚至需要脱去保护重新回收原料。
一些常用的酸性条件:
- 100 mol% 10-CSA(樟脑磺酸)的甲醇溶液,室温;反应十分钟可脱除一级的TBS保护基。
- 10 mol% 10-CSA,1:1体积比的甲醇:二氯甲烷,−20或0°C;0摄氏度下反应2个小时脱除一级TBS保护基;若使用PPTS(对甲苯磺酸吡啶盐)代替CSA,则速率降低十倍;而使用pTsOH(对甲苯磺酸)则速率提高10倍。
- 4:1:1(v/v/v)乙酸:四氢呋喃:水的溶剂体系下室温反应,反应速率很慢但选择性很好。
一些常用的碱性条件:
参考文献
- Corey, E. J.; Venkateswarlu, A. "Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives." J. Am. Chem. Soc. 1972, 94, 6190–6191. doi:10.1021/ja00772a043
- Corey, E. J.; Cho, H.; Rücker C.; Hua, D. H. "Studies with trialkylsilyltriflates: new syntheses and applications." Tetrahedron Lett. 1981, 22, 3455–3458. doi:10.1016/S0040-4039(01)81930-4
- Greene, T. W.; Wuts, P. G. M. Protective Groups In Organic Synthesis, 3rd ed.; John Wiley & Sons: New York, 1991.
- Greene, T. W.; Wuts, P. G. M. Protective Groups In Organic Synthesis, 3rd ed.; John Wiley & Sons: New York, 1991.
- McDougal, P. G.; Rico, J. G.; Oh, Y.-I.; Condon, B. D. "A convenient procedure for the monosilylation of symmetric 1,n-diols." J. Org. Chem. 1986, 51, 3388–3390. doi:10.1021/jo00367a033
- Roush, W. R.; Gillis, H. R.; Essenfeld, A. P. "Hydrofluoric acid catalyzed intramolecular Diels-Alder reactions " J. Org. Chem. 1983, 49, 4674-4682. doi:10.1021/jo00198a018
- Hu, L.; Liu, B.; Yu, C. Tetrahedron Lett. 2000, 41, 4281. doi:10.1016/S0040-4039(00)00626-2
- Holton, R. A. et al. "First total synthesis of taxol. 2. Completion of the C and D rings." J. Am. Chem. Soc. 1994, 116, 1599–1600. doi:10.1021/ja00083a067
- Nelson, T. D.; Crouch, R. D. "Selective deprotection of silyl ethers." Synthesis 1996, 1031–1069. doi:10.1055/s-1996-4350
- Crouch, R. D. "Selective monodeprotection of bis-silyl ethers." Tetrahedron 2004, 60, 5833–5871. doi:10.1016/j.tet.2004.04.042
- Higashibayashi, S.; Shinko, K.; Ishizu, T.; Hashimoto, K.; Shirahama, H.; Nakata, M. "Selective deprotection of t-butyldiphenylsilyl ethers in the presence of t-butyldimethylsilyl ethers by tetrabutylammonium fluoride, acetic acid, and water." Synlett 2000, 1306–1308. doi:10.1055/s-2000-7158