陽離子聚合反應
陽離子聚合反應為一種鏈增長聚合反應,反應中陽離子引發劑(cationic initiator)會將電荷轉移單體,使單體變得有反應性。而這個具反應性單體會與其他單體進行相似的反應,最後形成聚合物。.[1][2] 陽離子聚合反應中的這種單體,必須受具有提供電子取代基(electron-donating substituent)的烯烴(olefins)和雜環(heterocycles)限制。相似於陰離子聚合反應,陽離子聚合反應對反應中所使用的溶劑(solvent)非常敏感,特別是溶劑可形成游離離子(free ions)的能力,此能力決定了增長的陽離子鏈的反應性。 陽離子聚合反應用於聚異丁烯(polyisobutylene)的產生(如:丁基橡膠用於輪胎的內胎)和聚N-乙烯基咔唑(N-vinylcarbazole,PVK)[3]
單體
陽離子聚合反應的單體規模受限於兩個主要種類:烯烴(Olefins)和具雜環單體(Heterocyclic monmers)。若要兩種單體同時發生陽離子聚合反應,只有在利於熱(thermally favorable)的反應中才會發生。以烯烴單體為例,是因為此單體中的雙鍵之異構化反應(isomerization);而對雜環單體來說,是因為單體之環張力(ring strain)的釋放和在有些例子中,重複的單元的異構化反應。陽離子聚合反應中的單體具親核性(nucleophilic),且在聚合反應中可形成穩定的陽離子。[5]
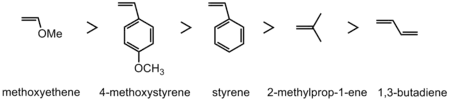
烯烴
具有提供電子的取代基的烯烴單體會發生陽離子聚合反應,這些提供電子的取代基使烯烴具有親核性,足以攻擊親電子性引發劑(electrophilic initiators)或使聚合鏈增長。同時,這些接上單體之提供電子的取代基能穩定對接下來聚合反應所產生的陽離子電荷。有些具有反應性的烯烴,具有雜原子基(heteroatom groups)的反應性會好於烷基(alkyl)或芳香基(aryl groups),在下圖從左至右反應性越差為排序,表現烯烴的反應性。無論如何,碳烯陽離子(carbenium ion)形成的反應性與單體的反應性相反。[5]
具雜環單體
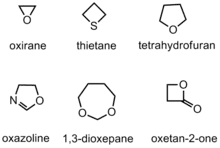
具雜環單體會陽離子聚合成內酯(lactone)、內醯胺(lactam)和環胺類(cyclic amine),當加入引發劑(initiator)時,環狀單體會產生線性聚合物(linear polymers)。具雜環單體的反應性取決於,其自身的環張力,具有大環張力的單體,如環氧乙烷(oxirane),環氧乙烷相較於1,3-二氧環庚烷(1,3-dioxepane)具有較大的反應性,因為1,3-二氧環庚烷的環張力較小。六個(或以上)碳形成的環不傾向發生聚合反應,因為環張力較低。[6]
合成
链引发
链引发為陽離子聚合反應的第一步驟,在链引发步驟中會從已做好的聚合鏈中產生碳烯陽離子(carbenium ion)。抗衡離子(counterion)應為非親核性的(non-nucleophilic),否則反應會瞬間終止。對陽離子聚合反應有許多引發劑(initiators),有些引發劑需要共引發劑(coinitiator)來產生所需種類的陽離子。[7]
典型的質子酸
強質子酸可使用於形成陽離子引發種類,若要產生足夠量的不同種陽離子,需要高濃度的質子酸。產生的抗衡離子(A–)具有較弱的親核性,因為要避免其與帶質子的烯烴結合,而使反應提早終止。[5] 常見的質子酸有磷酸、硫酸和三氟甲磺酸只有分子量小的聚合物可形成這些引發劑。[1]
路易斯酸/傅-克反應催化劑
路易斯酸(Lewis acids)為最常使用於陽離子聚合反應的起始步驟的化合物,其中常使用的路易斯酸為SnCl4、AlCl3、 BF3和TiCl4,雖然單獨使用這些路易斯酸可引起聚合反應,且此反應會與合適的陽離子來源快速發生。陽離子來源可為水、酒精,甚至或碳陽離子提供者(carbocation donor),如酯或酸酐。在此系統中,當碳源為引發劑時,路易斯酸作為共引發劑。當引發劑與共引發劑反應,反應中的中間複合物(intermediate complex)可與單體進行反應。由引發劑-共引發劑複合物產生的抗衡離子較路易斯酸A–親核性低。當加入反應性較好的路易斯酸時,鹵素(Halogens),如氯、溴也可引發陽離子聚合反應。[1]

碳烯陽離子鹽
只有具最好反應性的烯烴,可用穩定的碳烯陽離子來誘導烯烴鏈的增長,以得到明確的結構。在動力學中最常研究起始反應,因為測量碳正離子吸收值的消失是比較容易的。常見的碳烯陽離子有三苯基氯甲烷和鎓陽離子。[5]

游離輻射
游離輻射(Ionizing radiation)可形成自由基陽離子對,並與單體反應而開始了陽離子聚合反應。要控制自由基陽離子很困難,且受單體和反應環境影響。自由基的形成和陰離子的種類也常常是研究目標。[5]
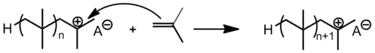
链增长
链增长(Propagation)的進行是透過加入單體給活化的種類(即碳烯離子)。單體是以從頭到尾(head-to-tail)形式加入至增長的鏈中;在這個過程,會產生陽離子的尾端,而下一個循環的單體則由此加入。[6]
溫度的影響
反應時的溫度會影響增生速率,整個陽離子聚合反應的活化能()取決於起始反應()、增生反應()和終止反應()的活化能。





常理來說,值小於和,這意味著整個反應的活化能為負值。但在這情況下,當反應溫度下降,使增生速率增加,相反地,這個反應的活動能為正值。[6]
反應溫度也影響了鏈的長度,低反應溫度(170-190 K)可聚合成最長鏈,[6]得終止反應和其他副反應(side reactions)的活化能較增長反應還要大。[5][6]當反應溫度增加時,終止反應的能量障礙被克服,而使聚合反應過程中產生較短鏈。[6]
溶劑與抗衡離子的影響
溶劑(solvent)與抗衡離子(又稱為反離子,gegen ion)對增生反應速率有重要的影響,抗衡離子與碳烯離子有不一樣的結合距離,範圍從共價鍵的形成、離子對的緊密結合(不分開)、溶劑分離離子對(solvent-separated ion pair)(部分分開)和游離離子(完全分離)。[1][6]
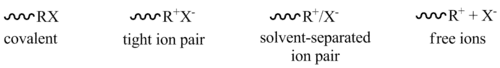
最強的交互作用為共價鍵的結合;最弱則是離子對中出現自由離子。[6]在陽離子聚合反應中,離子在離子對與自由離子間會傾向平衡(不論是緊密接合或溶劑分離時)。.[1]反應中使用的溶劑越具極性(polar),溶劑化(solvation)和離子的分離越好,因為自由離子的反應性較離子對高,其增生速率在極性溶劑中會比較快。[6][8]
抗衡離子的大小也是一個決定因素,較小的抗衡離子,具有較高的電子密度,與碳烯離子反應時也具有較強的靜電交互作用;較大的抗衡離子則電子密度較低。[1]近一步來說,一個較小抗衡離子與極性溶劑作用時,相較電子密度較低的抗衡離子來說,會比較容易溶劑化。這個結果表示,當增加溶劑的溶劑化能力時,可提高的增生速率。[6]
链终止
終止反應的發生是透過抗衡離子與單一分子的轉移,在這個過程中抗衡離子的陰離子部分會與增生鏈的末端結合。若減少引發劑-共引發劑複合物的濃度,這不只會讓增生鏈失去活性,也會使動力鏈(kinetic chain)被終止。[1][6]

鏈的轉移
有兩種方式能發生鏈的轉移:
1、從活性鏈(active chain)的末端分離氫(hydrogen abstraction),並轉移至抗衡離子上。[6][8][9]在此過程中,鏈的增長會終止,但是引發劑-共引發劑複合物(initiator-coinitiator complex)會產生,誘導更多鏈的產生。[5][6]
2、從活性鏈(active chain)的末端分離氫(hydrogen abstraction),並轉移至單體上。這個步驟會增長鏈終止,但也會形成新的碳烯陽離子-抗衡離子複合物(carbenium ion-counterion complex),此複合物會讓增長反應繼續,讓動力鏈(kinetic chain)維持不變。[6]
陽離子開環聚合反應
陽離子開環聚合反應(Cationic ring-opening polymerization)與起始、增生、終止反應所以走的反應機制相同。然而,在這個陽離子開環聚合反應中,單體單元需相較產物線狀聚合鏈成環狀。這個產物線狀聚合鏈可以降低上限溫度(ceiling temperature),因此聚合鏈的封端(end-capping),必須避免解劇作用(depolymerization)。[6]

動力學
增生反應速率和聚合化的程度可以由聚合反應的化學動力學來分析,起始、增生、終止反應和鏈轉移反應的反應方程式,以下為常見形式:




其中,I+為引發劑、M為單體、M+為增生中心(propagating center);、、和分別為起始、增生、終止反應和鏈轉移反應的反應速率常數。[5][6][10] 簡單來說,抗衡離子並未出現在以上反應方程式中,且只考慮鏈轉移反應的單體。以下為產生速率方程式(The resulting rate equations),括弧表示反應濃度。




![{\displaystyle \textstyle {\text{rate(initiation)}}=k_{i}[{\text{I}}^{+}][{\text{M}}]}](../I/4110013b7c2a9d796bf51b676934f36dd1014ee9.svg)
![{\displaystyle \textstyle {\text{rate(propagation)}}=k_{p}[{\text{M}}^{+}][{\text{M}}]}](../I/9096cb78356ba9d4bcf577b3aefa70f5f41d0b55.svg)
![{\displaystyle \textstyle {\text{rate(termination)}}=k_{t}[{\text{M}}^{+}]}](../I/c1d5aff19ead0a9e64481522c491fb98fd4e56f1.svg)
![{\displaystyle \textstyle {\text{rate(chain transfer)}}=k_{tr}[{\text{M}}^{+}][{\text{M}}]}](../I/534b33d2218447a6455f79614acf7e164f93d8fa.svg)
假設,在穩態條件下,即起始反應的速度=終止反應的速度:[6][10]
![{\displaystyle [{\text{M}}^{+}]={k_{i}[{\text{I}}^{+}][{\text{M}}] \over k_{t}}}](../I/21d942c4774e30d37953ab621eda557e80dcdcd9.svg)
這個方程式中的,[M+]可用於增生反應速率的方程式中:[6][10]
![{\displaystyle {\text{rate(propagation)}}={k_{p}k_{i}[{\text{M}}]^{2}[{\text{I}}^{+}] \over k_{t}}}](../I/104a8b28b7afdaff20ff360f7e1fef736cafe18e.svg)
從上述方程式中,可得,當增加反應單體和引發劑的濃度時,增生反應速率也會增加。
聚合化的程度(),可從增生反應速率和終止反應速率求得。:[6][10]

![{\displaystyle Xn={{\text{rate(propagation)}} \over {\text{rate(termination)}}}={k_{p}[{\text{M}}] \over k_{t}}}](../I/85e21127871e5988e2018b1e8534541f225047c5.svg)
若終止反應不如鍊轉移反應明顯,這的方程式會變成[6][10]

活性聚合反應
活性聚合反應(Living polymerization): 在1984年,東村(Higashimura)和澤本(Sawamoto)提出對烷基乙烯基醚(alkyl vinyl ethers)的第一個活性陽離子聚合反應。這個種類的聚合反應,受明確的單體控制;在活性陽離子聚合反應中,終止反應扮演了重要的角色,因為終止反應是基本消除(essentially eliminated),故陽離子鏈會持續增長,直到單體被消耗殆盡[11]。
商業應用
陽離子聚合反應最大的商業應用是,製造聚異丁烯(polyisobutylene,PIB)產物,包含聚丁烯(polybutene)和丁基橡膠(butyl rubber)。這些聚合物有各式各樣的應用,從黏合劑(adhesives)和密封剂(sealants)到防護手套(protective gloves)和醫療瓶塞(pharmaceutical stoppers)。合成各式的異丁烯產物的反應環境,取決於產物欲產生的分子量和使用的單體種類。最常用到的反應條件為,低分子量(5–10 x 104 Da)的聚異丁烯或TiCl4處於溫度範圍為−40 to 10 °C。[1]這些低分子量的聚異丁烯聚合物可用於填補縫隙,如密封剂(sealants);[1]高分子量的PIB會在較低溫度範圍(−100 to −90 °C)下、且在二氯甲烷(methylene chloride)的極性介質內合成。[5] 這些聚合物可用於,產生未相連的橡膠產物和熱塑性塑料(thermoplasts)的添加物;而其他高分子量PIB的特性為低毒性,使其可作為口香糖的基底。產生聚異丁烯之主要的化學公司,如埃索(Esso)、艾克森美孚(ExxonMobil)、巴斯夫(BASF)。[12]

丁基橡膠(Butyl rubber)與PIB相反,丁基橡膠為共聚物(copolymer),而其單體有~98%為異丁烯(isobutylene)和2%的異戊二烯(isoprene),其具核過程相似於高分子量PIB的聚合過程。丁基橡膠聚合反應會在AlCl3作為引發劑的情況下持續產生,它的低透氣性(gas permeability)、對化學物質的抵抗力(resistance)和抵抗老化,使其可各式應用,如防護手套、電纜絕緣體(electrical cable insulation),甚至籃球。在第二次世界大戰時期,丁基橡膠的產品越來越多,將近現今美國產生十億 磅/年產品。[1]
聚丁烯為另一種共聚物,包含近80%異丁烯和20%其他丁烯(通常為1-丁烯),這些低分子量聚合物的產生(300-2500 Da),可在較大的溫度範圍(−45 to 80 °C)中與AlCl3或BF3產生。以這些聚合物的分子量來看,它們可作為黏合劑、密封剂、塑化劑(plasticizers)和變速箱油(transmission fluids)的添加物,等等許多應用。這些物質為低花費且由不同的公司產生,包含BP化學、埃索(Esso)和巴斯夫(BASF)。[5]
其他由陽離子聚合反應形成的聚合物為均聚物(homopolymers)和聚萜烯(polyterpenes)的共聚物(copolymers),如植物衍生物蒎烯(pinene),可作為增黏劑(tackifiers)。在雜環(heterocycles)領域下,1,3,5-三氧雜環己烷(1,3,5-trioxane)是與少量環氧己烷(ethylene oxide)共聚合,而成的高結晶性(highly crystalline)聚甲醛(polyoxymethylene)塑料;烷基乙烯基醚(alkyl vinyl ethers)的均聚合作用(homopolymerization)只能以陽離子聚合反應達到。[1]
參考
- Odian, George. 4th. Hoboken, NJ: Wiley-Interscience. 2004. ISBN 978-0-471-27400-1.
- Mark, Herman F.; Bikales, Norbert; Overberger, Charles G.; Menges, Georg; Kroschwitz, Jacqueline I. 2nd. Wiley-Interscience. 1990. ISBN 978-0-471-80950-0.
- Robello, Douglas R. . Department of Chemistry, University of Rochester. 2002 [20 March 2011]. (原始内容存档于2011-07-20).
- Jenkins, A. D.; Kratochvíl, P.; Stepto, R. F. T.; Suter, U. W. (PDF). Pure and Applied Chemistry. 1996, 68 (12): 2287–2311 [2014-06-24]. doi:10.1351/pac199668122287. (原始内容存档 (PDF)于2016-03-04).
- Matyjaszewski, Krzysztof. . New York, New York: Marcel Dekker, Inc. 1996. ISBN 978-0-8247-9463-7.
- Cowie, John M. G.; Arrighi, Valeria. 3rd. Boca Raton: Taylor & Francis. 2008. ISBN 978-0-8493-9813-1.
- Kennedy, J. P.; Marechal, E. . Journal of Polymer Science: Macromolecular Reviews. 1981, 16: 123–198. doi:10.1002/pol.1981.230160103.
- Raave, A. 2nd. New York, New York: Kluwer Academic/Plenum Publishers. 2000. ISBN 978-0-306-46368-6.
- Fahlman, Bradley D. . Springer. 2008 [2014-06-25]. ISBN 978-1-4020-6119-6. (原始内容存档于2017-04-23).
- Ebewele, Robert Oboigbaotor. . Boca Ration, FL: Chapman & Hall/CRC Press LLC. 2000 [2014-06-25]. ISBN 978-0-8493-8939-9. (原始内容存档于2017-04-23).
- Sawamoto, M. . Progress in Polymer Science. 1991, 16: 111–172. doi:10.1016/0079-6700(91)90008-9.
- Chanda, Manas; Roy, Salil K. 4th. Boca Raton, FL: CSC Press. 2007 [2014-06-25]. ISBN 978-0-8493-7039-7. (原始内容存档于2017-04-23).