活性聚合反應
活性聚合反應在高分子化學中是一種鏈增長聚合反應,然而在活性聚合反應中,聚合物鏈的活性端並不具有鏈終止的能力。[1][2]這種聚合反應中缺少鏈終止反應和鏈轉移反應,且聚合起始的速度遠比鏈成長的速度來得大,以上特性產生的結果是:聚合物鏈生長的速度比傳統聚合方法的速度還來得恆定,且這些鏈的長度是相似的(即它們具有非常低的多分散性指數)。活性聚合反應對於區段共聚物的合成是一種常用的方法,由於可以分階段進行合成,所以可以控制不同階段合成的鏈段上具有不同的單體。另外的優點是可以預定想合成的聚合物的分子量,及控制末端基團。
, 使動能鏈所帶的能量在聚合反應中是穩定的。[3]
活性聚合反應是理想的聚合反應,因為它為聚合物的合成提供了精準度及可控性──由於聚合物的特性常常表現在他們的微觀結構以及分子量上,這兩點非常重要。相較之下,要在非活性聚合反應中,對分子量和多分散性指數的可控性是比較低的。[4][5]
在許多情況下,活性聚合反應會被混淆或認為是控制聚合反應。雖然這兩個反應非常相似,但還是有明顯的區別來區分這兩個反應。活性聚合反應被定義為終止或鏈轉移被移除的聚合反應,可控制聚合反應終止的地方被抑制,但不被移除,藉此來引發聚合物的休眠狀態。然而,這區分仍然是一個有爭議的文獻。
主要的活性聚合反應技術是:
- 活性陰離子聚合反應
- 活性陽離子聚合反應
- 活性開環易位聚合反應
- 活性自由基聚合反應
- 活性鏈增長縮聚反應
歷史
活性聚合反應是由邁克爾史瓦克在1956年首先證明。他以苯乙烯作為單體,以鹼金屬 / 萘的系統作為起始劑,在四氫呋喃(THF)中進行陰離子聚合反應。他發現,在起始劑系統中加入單體後,黏度便會增加(聚合物的分子量上升時會增加其溶液的黏度,因此黏度可作為聚合反應進行與否的一個指標),當單體被消耗完後,這個黏度增加的現象就隨之停止。然而,他發現在此時再次加入額外劑量的單體,居然可使反應系統的黏度繼續增加。這個現象代表了聚合物鏈的成長,並導出了一個結論──聚合物鏈的活性端始終保持著活性,且聚合反應從未被終止。[6]這個發現是高分子化學領域裡一個重要的里程碑,因為在一般情況下,聚合時的淬火或終止反應通常是我們無法控制的。有了這個發現,使本領域之潛在應用的研究蓬勃發展了起來。在今日,活性聚合反應廣泛應用於多種不同的聚合物或塑料的開發。藉由使用活性聚合,現代的化學家可以很容易地控制聚合物的化學組成,乃至於該材料的結構和電性質等等。[7]
活性聚合反應中最常見的類型包括:陰離子聚合,陽離子聚合,自由基聚合,或開環反應等等,每一種聚合反應的具體細節都會在下面的章節進行介紹。
特性
活性聚合的特徵
活性聚合的主要特徵是,鏈終止反應和鏈轉移反應基本上是由鏈增長聚合反應中的起始反應和增長反應來移除。
快速的起始速度
另一個關鍵特徵是起始的速度(表示休眠化學物種產生活性鏈增長物種)必須比鏈增長的速率要快得多。這表示了活性物種在鏈延長反應時會以相同速度成長。相反來說,鏈起始速度如果比鏈延長速度來得慢(起始反應是速率控制步驟),則活性物質會導致各個聚合物鏈之間的分佈較寬,而在反應過程中形成的不同的聚合度(或鏈長度)和分子重量。
低分散度
分散度(DJ)或是多分散性指數(PDI)是廣度的聚合物鏈分佈,由於沒有鏈終止路徑,生物聚合物趨向於具有低Đ,還有起始速率比延長速率來得快。如果鏈終止存在,則鏈將“死”或導致聚合物鏈具有不同Xn,聚合過程中在不同的時間缺少活性。如前一節中所述,如果延長速度比起始速率要慢得多,那麼所有的起始劑將會在開始延長前形成活性物種。這兩個特徵是顯而易見的,活性鏈的濃度,也就是那些發生聚合反應,都變成一種基本恆定。這兩個特徵擴展了延長鏈的壽命,包括允許合成操縱,共同嵌段聚合物的形成,還有在活性鏈中進行端基官能化。增加延長聚合物鏈的壽命,可允許對於聚合物的結構和性能的控制,因為聚合物的結構與性能具有一定的關係。
可預測的分子量由於活性聚合反應中缺少鏈終止和鏈轉移,則各起始劑會各產生一個活性物種,一個活性物種負責一條鏈。這提供了平均聚合度的控制,這是關係到Mn的平均度控制(數均分子量),通過控制單體([M]o)與引發劑([I]o)的比值。在理想的活性系統下,假定產生活性物種的效率是100%,其中,每個引發劑只產生一種活性物質的動力學鏈長(活性物種單體的平均數在其壽命期間的反應),在一定時間裡可以通過以下方式來計算剩餘的單體濃度。數均分子量,Mn,在活性聚合反應下,與轉化率呈線性增加。
![{\displaystyle \ v={\frac {[M]_{0}-[M]}{[I]_{0}}}}](../I/db304320640f163330c3130db03ff9691e1a8ffa.svg)
活性聚合技術
活性陰離子聚合反應
早在早在1936年,卡爾·齊格勒提出,在苯乙烯和丁二烯連續加入單體,以烷基鋰引發劑引發陰離子的聚合反應不會發生鏈轉移或終止。二十年後,通過活性聚合反應證明了史瓦克的陰離子聚合中的苯乙烯在THF用鈉萘基作為催化剂。[8][9][10]
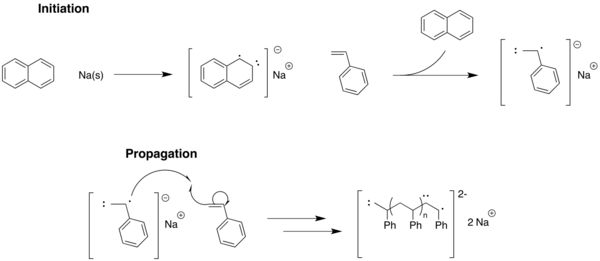
在這裡,萘陰離子作為聚合反應的引發劑來活化苯乙烯。但是,請注意(無雜質淬火和無溶劑鏈轉移)沒有發生終止的途徑。這是基礎陰離子活性聚合反應,其中末端基團會存在,直到游離單體可用於額外的延長,或從外部淬滅。
活性α-烯烴聚合反應
α-烯烴可經由陰離子配位中心聚合反應進行聚合,催化劑的金屬中心被認為是抗衡陽離子的烷基鏈末端(經由MR協調)。齊格勒-納塔起始劑在50年代中期被開發,且非均相起始劑被使用在α-烯烴的聚合。這些起始劑的不僅第一個實現分子量相對高的聚(1 -烯烴)(目前在世界上最廣泛生產的熱塑性塑料是PE(聚乙烯)和PP(聚丙烯)[11],起始劑也能有立體選擇性聚合反應,這歸因於該手性晶體結構的多相起始劑。由於這個發現的重要性,使齊格勒和納塔在1963年得到諾貝爾化學獎。雖然來自齊格勒-納塔起始劑形成的活性物質一般具有長壽命(幾個小時或更長的時間),由於多種鏈轉移途徑(β-氫消除反應,並轉移到共起始劑)使延長鏈的壽命被縮短,最後不視其為活性物。
金屬茂起始劑被認為是齊格勒-納塔起始劑的一種,由於使用了雙組分體系的過渡金屬和一組I-III族金屬的共起始劑(例如甲基鋁氧烷(MAO)或其它烷基鋁化合物)。茂金屬起始劑形成了均勻的單中心催化劑,最初的開發研究表示催化劑的結構對於聚合物結構/性能所產生的影響。[12]各個不同的研究人員對茂金屬催化劑能夠調整並涉及如何輔助配體(沒有直接參與化學變化)結構和影響聚合物微觀結構的對稱手性金屬中心。[13]然而,我們很難去知道斷鏈反應(主要是β-氫消除反應)中的茂金屬基的聚合。
通過調整輔助配體及取代基起始劑的空間位阻和電子性質,被稱為螯合物起始劑(或後茂金屬起始劑)已被成功地用於α-烯烴的立體專一活性聚合反應中。螯合起始劑具有高潛力的活性聚合能力,因為輔助配體可以被設計為阻止或抑制鏈終止。螯合起始劑在輔助配體下可進一步細分為; 柄型- 環戊二烯基-酰氨基的引發劑,α-二亞胺螯合物和苯氧基-亞胺螯合物。
- 柄型 - 環戊二烯基 - 酰胺(CPA)的起始劑
CPA的起始劑具有一個環戊二烯基的取代基和一個或多個氮的取代基配位到金屬中心(通常是Zr或Ti)。甲基(五甲基環戊基)鋯乙脒在-10℃下與1 - 己烯被用於立體專一活性聚合反應。從結果來看,13 C-NMR證實了聚(1 - 己烯)為全同立構物(相鄰的重複單元之間有相同的立體化學)。多次試驗顯示M n跟低的Đ是可控和可預測的(從催化劑與單體的比例)。聚合反應進一步證實能使此帶活性且依次添加2個單體,在第一個單體被聚合後第二個單體就被加上去,並且監視鏈的 Đ 和 M n值。
- α-二亞胺螯合起始劑
α-二亞胺螯合起始劑的特徵在於具有一個二亞胺螯合輔助配體的結構和它一般配位到過渡後期(即鎳和鈀)的金屬中心。
布魯克哈特等。對於該類催化劑有廣泛的研究和報導活性聚合α-烯烴[14]和證實活性α-烯烴的一氧化碳的交替共聚物[15]。
- 苯氧基 - 亞胺螯合物
活性陽離子聚合反應
單體對於陽離子聚合反應時是富電子烯烴的,如乙烯基醚,異丁烯,苯乙烯和N-乙烯基咔唑。起始劑是二進位系統,包括一個親電子和路易斯酸。該方法在1980年左右被開發,歸功於東村,澤本和肯尼迪。在正常情況下,在延長的時間週期裡產生穩定的碳正離子是困難的,由於有可能陽離子被β-質子淬滅,連接到另一單體的主鏈中,或者是一個自由的單體。所以我們採取不同的方法[16][17]
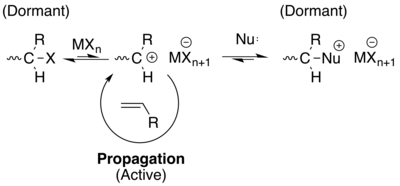
在這個例子中,碳陽離子被加入一種路易斯酸(見圖共起始劑,以及已經在聚合物中的鹵素的“X”),最終會產生一個弱平衡的碳陽離子。此平衡傾向於休眠狀態,以利其他途徑的進行。此外,加入弱親核試劑可以進一步降低活性物質的濃度,保持聚合物的活性。[16][17]然而,值得注意的是,根據定義,在這個例子中所描述的聚合物不是技術上的活性,由於引入了休眠狀態,終止只被減少,不排除(雖然這個話題還在爭議中)。但是,他們照常地使用這個方法,並且應用於類似的真實活性聚合反應。
活性開环易位聚合反應
在合適的反應條件下,開環易位聚合(ROMP)就可以產生活性。由羅伯特·格拉布在1986年對降冰片烯和特伯試劑此類系統進行了描述,並在1978年格拉布斯以及理察·施羅克描述鎢碳烯複合物的活性聚合反應。[18]
通常,此反應涉及轉換環狀烯烴的環(> 5千卡/莫耳),如環丁烯,降冰片烯,環戊烯等,聚合物也含有雙鍵。環易位聚合反應需要注意的是雙鍵通常保持在主鏈,y在適當條件下可被認為是有活性的。[19]
對於ROMP反應來說,滿足以下條件者,可被視為有活性[19]
- 單體的起始反應是快速且完整的。這表示起始劑活化單體的速度是非常快的。
- 各聚合物(聚合度)有多少單體組成與你開始使用的量呈線性關係
- 聚合物的分散度必須<1.5。換句話說,要知道你的聚合物鏈有多長,分布要相當低才行。
有了這些條件後,就可以創造一個聚合物,可以控制它的內容(用什麼單體)和它的特性(大部分與長度有關)。要注意的是活性開環聚合反應可以是陰離子也可以是陽離子。

因為活性聚合物的終止能力被去除掉,這表示,一旦你的單體已被消耗時,可以加入更多的單體使該聚合物鏈繼續增長,直到所有單體被消耗殆盡。這將繼續下去,直到末端鏈的金屬催化劑將加入的焠火劑去除。就結果來看,它可以創造一個共聚物而且相當容易及準確。可以經由調整聚合物的性能來達到期望的應用(電子/離子導電等) [17][19]。
活性自由基聚合反應
從1970年代開始發現了一些新的方法,可以使用活性聚合反應來發展自由基化學。這些技術涉及催化鏈轉移聚合反應,引發轉移終止劑介導的聚合反應,穩定的自由基介導聚合(SFRP),原子轉移自由基聚合(ATRP),可逆加成-斷裂鏈轉移(RAFT)聚合和碘轉移聚合。
在“活性”自由基聚合(或受控的自由基聚合(CRP))下,相比傳統的自由基聚合(RP)和CRP可以顯示活性聚合的特而徵斷鍊途徑是被抑制的。然而,由於鏈終止不會消失,但會有最小化的情況,CRP在技術上不符合IUPAC規定的活性聚合要求(見IUPAC的定義)。這個問題尚未在文獻中解決,因此它經常被認為是一個“活”的聚合反應,準活性聚合反應,偽活性聚合反應和其他方面來代表這個問題。
CRP有兩個基本的策略,一是抑制斷鍊反應,二是促進起始反應比延長反應快。這兩種策略的基礎是建立於開發一個動態的平衡中積極延長激發態和休眠種。[20]
第一種策略涉及一種可逆誘捕機制,在其中延長的自由基與一個物種X經由激活/去激活(即原子轉移自由基聚合)的過程。其中物質X是一種持久性基團,或一種可以產生穩定的激態,即不能終止本身或延長,但能可逆“終止”與增長自由基(來自增長的聚合物鏈)P*。P*與另一個P*為一個激進物種的延長(Kp)和不可逆終止(kt)。X通常是氮氧化物(即TEMPO中使用氮氧介導的自由基聚合)或有機金屬物種。休眠物種(Pn-X)可以被激活,使用催化劑和光,可以自發地產生活性物種(P*)。[20][21]
第二個策略是在轉移劑之間延長激態,基於一種退化性轉移(DT),充當為休眠種(即可逆加成斷裂鏈轉移聚合)。DT基於CRP遵循常規反應動力學的自由基聚合,即慢啟動和快速終止,但轉移劑(Pm-X or Pn-X)的濃度比自由基起始劑的濃度來得高。延長自由基物種與休眠物轉移劑經由原子轉移,基團轉移或增加化學片段產生熱中性交換。[20]
活性鏈生長縮聚反應
鏈增長縮聚聚合反應在最初開發的前提下,是聚合物的取代基變化的效應,相對於單體,聚合物的端基有更多活性,被稱為“活性中間體縮聚”。主要的結果是單體與活化的聚合物末端基團的反應比與其它單體優先。這個優先的反應性是基本的差異用來分類聚合機制,作為鏈增長,而不是逐步增長,其中,單體和聚合物鏈的端基具有相等的活性(反應性是不受控制的)。對於小分子量的聚合物,有多種策略被用來最小化單體-單體反應(或自縮合),聚合低D和可控制的Mn可以使用此種機制。[22]然而,對於高分子量的聚合物鏈(即小的引發劑與單體的比)的Mn是不容易控制的,對於一些單體來說,由於增長物種濃度低,所以單體之間自縮合的頻率更高。[22]
參考文獻
- Halasa, A. F. Rubber Chem. Technol., 1981, 54, 627.
- (2006) The Chemistry of Radical Polymerization - Second fully revised edition (Graeme Moad & David H. Solomon). Elsevier. ISBN 0-08-044286-2
- . Pure and Applied Chemistry. 1996, 68 (12): 2287–2311. doi:10.1351/pac199668122287.
- Cowie, J.M.G. (2007). Polymers chemistry and physics of modern materials (3rd ed / J.M.G. Cowie and Valeria Arrighi ed.). Boca Raton: Taylor & Francis. ISBN 9780849398131.
- Odian, George (2004). Principles of polymerization (4. ed. ed.). Hoboken, NJ: Wiley-Interscience. ISBN 0471274003.
- Webster, O. W. Science, 1991, 251, 8877.
- McNeil, Anne; Bryan, Zachary (2013). "Evidence for a preferential intramolecular oxidative addition in Ni-catalyzed cross-coupling reactions and their impact on chain-growth polymerizations". Chem. Sci. 4: 1620–1624. doi:10.1039/C3SC00090G.
- M. Szwarc, Nature 1956, 178, 1168.
- Szwarc, M.; Levy, M.; Milkovich, R. J. Am. Chem. Soc. 1956, 78, 2656.
- US 4 158 678 (priority date 30 June 1976).
- Craver, C., Carraher,C. . Elsevier. 2000: 1022–1023.
- Craver, C., Carraher,C. . Elsevier. 2000: 1022–1023.
- Coates, Geoffrey W. . Chemical Reviews. April 2000, 100 (4): 1223–1252. doi:10.1021/cr990286u.
- Killian, C.; Tempel,D;Johnson,L.; Brookhart, Maurice. J. Am. Chem. Soc. 1996, (118): 111664–11665. 缺少或
|title=为空 (帮助) - Brookhart, Maurice; Rix, Francis; DeSimone, J. J. Am. Chem. Soc. 1992, (114): 5894–5895. 缺少或
|title=为空 (帮助) - Cowie, J.M.G. 3rd ed / J.M.G. Cowie and Valeria Arrighi. Boca Raton: Taylor & Francis. 2007. ISBN 9780849398131.
- Odian, George. 4. ed. Hoboken, NJ: Wiley-Interscience. 2004. ISBN 0471274003.
- "Ring-opening polymerization of norbornene by a living tungsten alkylidene complex" R. R. Schrock, J. Feldman, L. F. Cannizzo, R. H. Grubbs Macromolecules; 1987; 20(5); 1169–1172. doi:10.1021/ma00171a053
- Bielawski, Christopher W.; Grubbs, Robert H. . Progress in Polymer Science. January 2007, 32 (1): 1–29. doi:10.1016/j.progpolymsci.2006.08.006.
- Braunecker, Wade A.; Matyjaszewski, Krzysztof. . Progress in Polymer Science. January 2007, 32 (1): 93–146. doi:10.1016/j.progpolymsci.2006.11.002.
- Matyjaszewski. . (原始内容存档于2014-03-14).
- Yokozawa, Yokoyama, T., A. Prog. Polym. Sci. 2007, 32: 147–172. 缺少或
|title=为空 (帮助) - Miyakoshi, Ryo; Yokoyama, Akihiro; Yokozawa, Tsutomu. . Journal of the American Chemical Society. December 2005, 127 (49): 17542–17547. doi:10.1021/ja0556880.