药物代谢动力学
药物代谢动力学,简称为:药代动力学(源自古希腊语 「药物」和 “移动,投入运动”;参见化学动力学),属于药理学的一个分支,致力于研究物质进入生物体内,随时间在体内变化的规律。学科研究的物质包括任何化学外源性物质,如:药物、农药、食品添加剂和化妆品等。药代动力学需要分析化学物质的代谢,并研究某化学物质从进入人体至完全从体内清除的过程及变化规律。药代动力学研究生物体如何影响药物在体内的行为,而药效学(Pharmacodynamics,PD)则研究药物如何影响生物体。如PK/PD模型所示,两者共同影响给药剂量、药物获益和药物副作用,两者通常同时进行研究,简称为PKPD。[1]
广义上的药代动力学包括了:药物代谢(Drug metabolism,DM)与药物动力学(Pharmacokinetics,PK),两者通常紧密相关被统称为药物代谢动力学(DMPK)。[1]
概述
药代动力学研究特定的外源性化学物质如何通过体内的吸收、分布和代谢机制(如通过细胞色素P450或葡萄糖醛酸转移酶等代谢酶)影响机体,并形成药物的代謝產物和排泄途径。[2]化合物的药代动力学特性受给药途径和给药剂量的双重影响,并一同影响吸收速率。[3]
在研究中,为简化生物体与化学物质相互作用的过程,可以运用某些模型研究药代动力学,如多隔室模型(或称多房室模型,多室模型,Multi-compartmental model)。建模类型中有些较为复杂,其中“单室模型”(Monocompartmental models)和”两室模型”(Two compartmental models)较为简单且常用。[4]模型中分隔出为不同的室通常称为ADME图(ADME scheme),若将释放过程(Liberation)从吸收过程中单独列出,也称为LADME:
- Liberation 释放——药物从制剂中释放的过程。[5][6]参见IVIVC 。
- Absorption 吸收 ——物质进入血液循环的过程。
- Distribution 分布——物质在身体体液和组织内的分散或传播。
- Metabolism 代谢——生物体识别存在外源性化合物,并将化合物不可逆地转化为代谢产物。
- Excretion 排泄——将物质从体内排出。一些药物若不可逆地在身体组织中积聚,可能会导致毒性。
药物的代谢和排泄两个阶段也可统称为药物消除(Elimination)。对以上药物在体内不同阶段的研究涉及一些基本概念,以了解药物体内的动态过程,其中诸多因素会影响到整个药代动力学过程,例如:赋形剂的物料属性对于释放产生影响;生物膜特有性质对于物质穿越膜及对药物吸收产生影响;酶反应的特性可导致药物失去活性等。
以上的概念均可通过相应数学公式来表达。通过理解药物分子的特性,以及药物基本物理化学性质,如酸解离常数(pKa)、生物利用度、溶解度、吸收能力和机体组织中的分布情况等,可推测其在有机体体内的药物动力学情况。
指标
| 参数 | 描述 | 符号 | 单位 | 公式 | 示例 值 |
|---|---|---|---|---|---|
| 剂量 | 服用药物量 | 设计参数 | 500 mmol | ||
| 服药间隔 | 服用药物间隔时间 | 设计参数 | 24 h | ||
| Cmax | 服药之后血药浓度峰值 | 直接测定值 | 60.9 mmol/L | ||
| tmax | 服药之后达到Cmax的时间 | 直接测定值 | 3.9 h | ||
| Cmin | 在下一次服药之前的最低血药浓度 | 直接测定值 | 27.7 mmol/L | ||
| Cavg | 在一个剂量间隔时间内,血药浓度达到稳态期间的平均血药浓度 | 55.0 h×mmol/L | |||
| 分布容积 | 某药物表观分布容积 | 6.0 L | |||
| 血药浓度 | 在一定体积血浆的药量 | 83.3 mmol/L | |||
| 吸收半衰期 | 一定剂量的药物吸收50%入体循环所需时间 | 1.0 h | |||
| 吸收速率常数 | 药物通过口服或血管外路径进入体内的速率 | 0.693 h−1 | |||
| 消除半衰期 | 药物在体内降低至原浓度一半时所需时间 | 12 h | |||
| 消除速率常数 | 药物从体内移除的速率 | 0.0578 h−1 | |||
| 输注速率 | 需要平衡药物消除的输注速率 | 50 mmol/h | |||
| 曲线下面积 | 一次性给药或在稳态时浓度-时间曲线的积分 | 1,320 h×mmol/L | |||
| 清除 | 每单位时间内清除药物血浆的体积 | 0.38 L/h | |||
| 生物利用度 | 药物在生物系统中可以利用的比例 | 无单位 | 0.8 | ||
| 波动 | 在稳态下一次给药间隔期间峰谷波动值 |
其中 |
41.8% |










































在药代动力学中,稳态(Steady state)是指药物总体的摄入量与其体内消除量处于动态平衡时的状态。实际情况下,一般开始规律性给药后,服药时间达到3至5倍半衰期后可达稳态。[1]
药物动力学模型
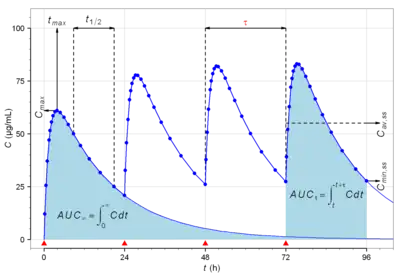
药代动力学通过非膈室或膈室方法进行建模。非隔室方法可通过估算血药浓度-时间图中的曲线下的面积(Area under the cruve,AUC)估计药物暴露量(Exposure)。隔室方法使用动力学模型估算血药浓度-时间图。非隔室方法不假设任何特定的隔室模型,而同样可准确预测生物等效性,因此被更广泛地使用。[4]药物在生物体内如何发生转化以及化合物最终的排泄过程,取决于许多相互关联的药代动力学因素。为了简化这方面的研究,已经开发了许多功能模型进行预测。这些模型基于将生物体视为许多相关隔室,如将有机体视为只有一个同类隔室。这种单室模型先假定药物的血浆浓度可真实反映药物在血浆体液或组织液中的浓度,并且药物的消除与生物体中药物浓度成正比,即一级动力学[10]。
然而,不存在完美准确反映体内的真实情况的模型。例如,并非所有身体组织具有相同的血液供给,因此药物在低血液供给组织中的药物分布会少于血液供应较好的组织。此外,有些如大脑的特定组织中存在的血脑屏障,对药物分布会构成较大障碍。若需要药物分布至有屏障的组织,必须有目的性地设计药物的相关特性以穿透相关屏障。若依据消除速率对组织进行区分,则生物体内可视为存在两类隔室:一类有更快的药物分布称为中央膈室,如主要脏器和血液供应发达的系统;另一类因其血流量较低称为外周隔室。其他组织如大脑,药物可能因血脑屏障而导致与其他器官组织分隔,可单独视为一个特殊膈室[11]。
药物消除过程发生在哪个隔室会导致隔室模型有所不同。最常见的情况是消除过程发生在中央隔室,如肝脏和肾脏因其血液供应丰富而最容易发生药物消除。然而,较少情况下消除可能发生在外周隔室或同时发生在两者中。这意味着两室模型中主要存在三种可能性。[12]
当负责代谢药物的酶(如细胞色素P450)发生饱和,或存在独立于血药浓度的主动消除机制,则经典药代动力学模型可能不适用。在实际情况中,每个组织都拥有独立的分布特性,药物在组织中不存在完全的线性关系。我们将药物在生物体中的分布容积定义为VdF,并将药物在组织中的分布容积定义为VdT ,则生物体的分布容积可由一个以下方程表述,其中考虑了不同作用类型的各种组织:[13][14]

多室模型可由多条药物浓度曲线组成,因此一条复杂的整体曲线需使用较复杂的数学方程表述。许多计算机程序已用于绘制这些方程和曲线。[12]无论模型可以设计得多复杂和尽量得精确,由于分布容积本质是为解决药代动力学问题而虚拟的一个简化概念模型,因此其预测结果与真实体内情况仍存在误差。为了尽量减少预测误差,找到与药物匹配模型非要必要。

非隔室模型分析
非隔室模型分析或称非房室模型分析(Noncompartmental Analysis,NCA),该分析法高度依赖对总药物暴露量的估算。总药物暴露常通过曲线下面积 (AUC) 方法估算,其中梯形法则(数值积分)是最常用的手段。梯形规则需要计算x的长度,因此面积估算结果高度依赖于血液/血浆采样时间计划。即取样时间点越密集,预测的梯形面积越能反映浓度-时间曲线的实际情况。NCA分析的结果准确性,取决于采样计划是否涵盖吸收、分布和消除的各个阶段,以准确表征药物的DMPK性质,如:AUC和暴露量、Cmax(最大浓度)、Tmax(最大浓度时间)、CL和Vd等参数。[15]
隔室模型分析
隔室PK分析使用动力学模型描述和预测浓度-时间曲线。PK隔室模型通常与其他学科中使用的动力学模型类似,如化学动力学和热力学。隔室分析优于某些非隔室分析的原因之一是能够随时预测浓度,缺点则是开发难度较高和需要适当的模型验证。最简单的PK室模型是单室PK模型,如静脉注射推注给药即是单室模型,并发生一级消除。最复杂的PK模型,如PBPK模型通过生理信息来简化模型开发和模型验证。[16]
单室模型
单室模型之所以称为线性药代动力学,是因为其涉及的各种药代动力学因素包括:剂量、血浆浓度、消除等,其浓度关系图可表达为线性关系或一个近似线性的关系。在线性药代动力学中为了使药物有效,药物需要从血浆快速转移至其他体液和组织中。[16]
浓度随时间的变化可表示为:

多室模型

多室模型即非药代动力学模型,其各因素之间呈非线性关系,因此其图例可用曲线表示,可通过计算曲线下不同区域的面积来评估各种因素之间的关联。非线性药代动力学中使用的模型主要基于Michaelis-Menten动力学。[17][18]
影响非线性药动力学的因素包括:
- 多相吸收:静脉注射的药物主要通过两种机制从血浆中清除:(1) 药物分布至身体各个组织 (2) 药物代谢+药物排泄。药物血浆浓度降低遵循双相模式(如图):[19]
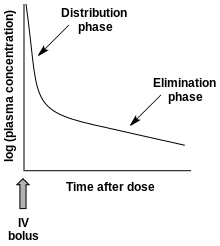 静脉给药后血浆药物浓度与时间的关系
静脉给药后血浆药物浓度与时间的关系 - 药物性质可决定高血流量和低血流量的组织分布情况。
- 酶饱和:药物代谢和转化需要特定生物酶,而当药物剂量达该酶处理能力阈值之上,则负责药物代谢的酶达到饱和。达到饱和点之后的血药浓度会不成比例地增加,即非线性。且药物的消除速率亦不再恒定。[22][23]
- 诱导或酶促抑制:一些药物具有抑制或刺激其自身代谢的能力,即处于负反馈或正反馈反应中。如药物氟伏沙明、氟西汀和苯妥英钠,随着这些药物剂量的增加,未代谢药物的血浆浓度会持续增加,而消除半衰期也延长,从而可能产生不良反应。因此,当需要高剂量用药时,必须调整剂量或其他治疗参数。[24]
- 肾脏还可为某些药物建立主动消除机制,而与血浆浓度无关。[25]
因此可以看出,吸收、分布、代谢和消除以上各个因素均可以影响整个药代动力学发生非线性过程。[26]
生物利用度

药物的生物利用度,也称生体利用率或生体可用率,在药理学上是指所服用药物的剂量部份能到达体循环的比例,是药物的一种药物动力学特性。[27][28][29]因此药物通过静脉给药因其直接进入人体循环,可提供最大的生物利用度,并且静脉给药被认为产生了 1(或 100%)的生物利用度,也称绝对生物利用度(Absolute bioavailablity,Fabs或BA)。使用其他药物递送方法的生物利用度与静脉注射的生物利用度,或特定研究中与其他递送方法相关的标准值(相对生物利用度)进行比较,即相对生物利用度(Relative bioavailability,Frel或BR)。[30][31]
![{\displaystyle B_{A}={\frac {[ABC]_{P}\cdot D_{IV}}{[ABC]_{IV}\cdot D_{P}}}}](../I/e0349b27d143a975c5e5c3eeaf83b49e8c7a3318.svg)
![{\displaystyle {\mathit {B}}_{R}={\frac {[ABC]_{A}\cdot {\text{dose}}_{B}}{[ABC]_{B}\cdot {\text{dose}}_{A}}}}](../I/634ea7e9316eeae72a9904dcaf1ccb2c43511aaf.svg)
一旦确定了药物的生物利用度,就可以基于需要达到的血药浓度调整药物剂量。因此,生物利用度是影响给药剂量的重要因素。可以使用以下公式计算血浆中真正可产生作用的药量:[32][33]

其中代表有效剂量, 代表生物利用度, 代表给药剂量。



如药物的生物利用度为 0.8(或 80%),且给药剂量为100毫克则:
- De = 0.8 × 100 毫克 = 80 毫克
即:每100 mg给药量将使得80 mg有效的进入血浆并影响药效。
生物利用度受每种药物固有的一系列因素影响,例如:[34]
这些概念可以通过数学量化和积分得到一个整体数学方程式:

其中是药物的纯度。[34]


其中是药物的给药速率,而是吸收的药物到达循环系统的速率。

最后,使用亨德森-哈塞尔巴尔赫方程并代入药物的酸度系数(电离药物与非电离药物之间存在平衡时的pH),可计算药物的非电离浓度,从而进一步计算吸收的浓度[35]:


LADME
一旦药物进入生物体随机发生药代动力学过程,这些过程可分为几个重要阶段,取其英文首字母缩写词为,LADME、ADMET或ADME,参见ADME[36]:
药物时常以活性形式给药,因此没有释放阶段或将释放与吸收阶段结合,并将分布、代谢和排泄结合为处置阶段(Disposition phase)。还可将药物毒理学方面研究结合ADME,称之为ADME-Tox或ADMET[37][38]。
以上每个阶段均受到药物和生物体之间的物理化学相互作用的影响,这些相互作用可以用数学模型进行研究。因此,药代动力学可表达为预测药物行为的数学方程式,且强调血药浓度与给药后时间两者的关系。
分析方法
生物分析方法
建立生物分析方法是构建浓度-时间曲线的必要过程。测试生物基质,如测量血浆中的药物浓度就使用化学技术进行。适当的生物分析方法应同时具备选择性与灵敏度,如微型热泳可用于量化生物基质/液体如何影响药物与其靶标的亲和力。[39][40]
质谱
基质如血样或尿样具有复杂性、虽然样品量较少并需长时间检验且维持高灵敏度,因此通常使用质谱法研究药代动力学。其中应用中最常用的仪器是配备三重四极杆质谱仪的LC-MS 。通常采用串联质谱法提高分析方法特异性。通常运用标准曲线和内标法进行样品中单一组分的定量分析。这些样本展示给药后,不同时间点的药物经代谢或从体内清除情况。给药前采集空白血液样本,对于确定测试背景和确保此类复杂样本的数据完整性非常重要。需要特别注意标准曲线的线性,当质谱数据在较宽泛的浓度范围下呈现非线性关系时,还可使用具有更复杂函数(例如二次函数)进行曲线拟合。[41][42][43]
目前科研工作者正在使用高灵敏度的质谱法进行微量剂量研究,这可能可以有效替代动物实验。[44]此外最新研究表明,二次电喷雾电离(SESI-MS) 可用于药物监测,可能进一步避免使用不必要的实验动物。[45]
群体药代动力学
群体药代动力学是研究个体药物浓度的变异性在群体中的来源和相关性,这些个体是接受临床相关剂量药物的目标患者群体。[46]某些患者的人口统计学、病理生理学和治疗属性,如体重、排泄和代谢功能以及是否同时接受其他治疗的信息,均改变剂量-浓度关系并可解释药物暴露量的可变性。例如,在肾功能衰竭患者中,主要由肾脏消除的药物稳态浓度通常高于接受相同药物剂量的肾功能正常患者。群体药代动力学旨在确定可测量的病理生理因素,并解释引起剂量-浓度关系变化的原因以及程度。若剂量-浓度关系变化影响到临床相关的治疗指数,并与血药浓度及药物暴露量变化直接联系,则必须适当修改剂量。群体药代动力学建模的优势在于能够分析较少的数据样本,当每位患者只有一个浓度测量值时仍可进行分析。[47][48]
临床药代动力学
| 抗癫痫药
药物 |
心脏活性
药物 |
免疫抑制剂
药物 |
抗生素
药物 |
|---|---|---|---|
|
|||
| 支气管扩张剂
药物 |
细胞生长抑制剂
药物 |
抗病毒药物
(艾滋病毒)药物 |
凝血因子 |
|
+依法韦仑
|
| |
临床药代动力学是源自群体药代动力学临床应用的一门学科,旨将有关药代动力学和患者人群特征的知识直接应用于临床治疗领域[49]。
环孢素药物作为免疫抑制剂的限制使用与重新启用,以帮助器官移植是一个相关案例。环孢素最初在临床中证明具有治疗效果,但发现许多用药患者出现肾毒性后其临床应用被严格限制。[50]然而,经过药代动力学的研究,例如通过分析患者的血浆浓度可以个体化指导患者环孢素的剂量。这种个体化用药使得环孢素可以再次广泛的应用于临床,并帮助了许多成功的器官移植手术。[51][52]
临床监测药代动力学参数,常通过测定血药浓度进行,因血液数据通常最易获取且较为可靠。[49][53]
在临床上出现以下情况需要监测药物血浆浓度:[54]
- 治疗窗口窄(致毒性浓度和药物起效浓度差)
- 高毒性
- 生命危险高
生态毒理学
生态毒理学是研究微塑料或其他对生物圈和环境有害物质的性质、影响以及相互作用的科学分支。[55]近些年发现造成环境污染的物质,如杀虫剂等可进入生物体并可能产生蓄积作用,因此药代动力学领域开展了对生态毒理学的研究。生产与生活中普遍应用的化学品均接受政府或国际机构(例如EPA或WHO)的研究和安全试验,以评估和阐述其对人体或生物体的影响。[56][57]这些化学物质在生物体内的滞留时间,即生物半衰期以及致死量等因素均是生态毒理学的主要关注点。[58]
参见
参考文献
- Gabrielsson J, Weiner D (2006) Pharmacokinetic and pharmacodynamic data analysis: concepts and applications, 4th edn. Swedish Pharmaceutical Press, Stockholm
- Pharmacokinetics. (2006).
- Knights K, Bryant B. . Amsterdam: Elsevier. 2002. ISBN 0-7295-3664-5.
- Levy, Gerhard, Milo Gibaldi, and William J. Jusko. "Multicompartment pharmacokinetic models and pharmacologic effects." Journal of pharmaceutical sciences 58.4 (1969): 422-424.
- Koch HP, Ritschel WA. . Landsberg, München: Ecomed. 1986: 99–131. ISBN 3-609-64970-4 (德语).
- Ruiz-Garcia A, Bermejo M, Moss A, Casabo VG. . Journal of Pharmaceutical Sciences. February 2008, 97 (2): 654–90. PMID 17630642. doi:10.1002/jps.21009.
- AGAH working group PHARMACOKINETICS. (PDF). Arbeitsgemeinschaft für Angewandte Humanpharmakologie (AGAH) (Association for Applied Human Pharmacology). 2004-02-16 [2011-04-04]. (原始内容 (PDF)存档于2016-05-08).
- Hallare, Jericho; Gerriets, Valerie, , StatPearls (Treasure Island (FL): StatPearls Publishing), 2021 [2021-12-25], PMID 32119385, (原始内容存档于2023-03-24)
- Pharmacokinetics Online Resource University of Lausanne Faculty of Biology and Medicine (FBM)
- Nestorov, Ivan A., et al. "Lumping of whole-body physiologically based pharmacokinetic models." Journal of pharmacokinetics and biopharmaceutics26.1 (1998): 21-46.
- Rescigno, Aldo. "Synthesis of a multicompartmented biological model." Biochimica et biophysica acta 37.3 (1960): 463-468.
- Milo Gibaldi, Donald Perrier.
- Biopharmaceutics and Pharmacokinetics Considerations Volume 1 in Advances in Pharmaceutical Product Development and Research 2021, Pages 195-277
- Handbook of Pharmacogenomics and Stratified Medicine 2014, Pages 341-364
- Benet LZ, Galeazzi RL (1979) Noncompartmental determination of the steady-state volume of distribution. J Pharm Sci 48:1071
- Mould, D. R., and Richard Neil Upton. "Basic Concepts in Population Modeling, Simulation, and Model‐Based Drug Development—Part 2: Introduction to Pharmacokinetic Modeling Methods." CPT: pharmacometrics & systems pharmacology 2.4 (2013): 1-14.
- International Union of Biochemistry (now International Union of Biochemistry and Molecular Biology). . Biochem. J. 1982, 213 (3): 561–571. PMC 1152169
 . PMID 6615450. doi:10.1042/bj2130561.
. PMID 6615450. doi:10.1042/bj2130561. - Cornish-Bowden, A. . Perspectives in Science. 2014, 1 (1–6): 74–87. Bibcode:2014PerSc...1...74C. doi:10.1016/j.pisc.2014.02.006.
- Gill SC, Moon-Mcdermott L, Hunt TL, Deresinski S, Blaschke T, Sandhaus RA. . Abstr Intersci Conf Antimicrob Agents Chemother. Sep 1999, 39: 33 (abstract no. 1195) [2023-05-10]. (原始内容存档于2011-11-23).
- International Union of Biochemistry (now International Union of Biochemistry and Molecular Biology). . Eur. J. Biochem. 1982, 128 (2–3): 281–291. doi:10.1111/j.1432-1033.1982.tb06963.x .
- Weiner D, Gabrielsson J. . . Apotekarsocieteten. 2000: 527–36 [2023-05-10]. ISBN 91-86274-92-9. (原始内容存档于2023-01-25).
- Stroppolo ME, Falconi M, Caccuri AM, Desideri A. . Cellular and Molecular Life Sciences. September 2001, 58 (10): 1451–1460. PMID 11693526. S2CID 24874575. doi:10.1007/PL00000788.
- Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, Tawfik DS, Milo R. . Biochemistry. May 2011, 50 (21): 4402–4410. PMID 21506553. doi:10.1021/bi2002289.
- Copeland RA. . Second. John Wiley & Sons, Inc. March 2013: 1–23. ISBN 978-1-118-48813-3. doi:10.1002/9781118540398.ch1.
- When Is Kidney Removal Necessary? Dr. Hunter Wessells, Hunter Wessells Reviewed: August 27, 2007
- International Union of Biochemistry (now International Union of Biochemistry and Molecular Biology). . Arch. Biochem. Biophys. 1983, 234 (2): 732–740. doi:10.1016/0003-9861(83)90262-X.
- Drug Bioavailability Gary Price Deven A. Patel In: StatPearls.Treasure Island(FL): StatPearls Publishing; 2023 Jan. PMID 32496732 Bookshelf ID: NBK557852 2022 Jun 23.
- Bioavailability: A Pharmaceutical Review 2011 J Novel Drug Deliv Tech
- Bioavailability L. Davidsson, S.A. Tanumihardjo, in Encyclopedia of Human Nutrition (Third Edition), 2013
- Roila, Fausto; Del Favero, Albano. . Clinical Pharmacokinetics. 1995-08-01, 29 (2): 95–109 [2023-05-13]. ISSN 1179-1926. doi:10.2165/00003088-199529020-00004. (原始内容存档于2023-05-13) (英语).
- Yang, Si H.; Lee, Myung G. . Biopharmaceutics & Drug Disposition. 2008-10, 29 (7): 414–426 [2023-05-13]. doi:10.1002/bdd.628. (原始内容存档于2023-05-13) (英语).
- Ondansetron clinical pharmacokinetics Clin Pharmacokinet. 1995 Aug;29(2):95-109.doi: 10.2165/00003088-199529020-00004.
- Dose-independent pharmacokinetics of ondansetron in rats: contribution of hepatic and intestinal first-pass effects to low bioavailability. Yang SH, Lee MG. Biopharm Drug Dispos. 2008 Oct;29(7):414-26.doi:10.1002/bdd.628.PMID 18697186
- Michael E. Winter, Mary Anne Koda-Kimple, Lloyd Y. Young, Emilio Pol Yanguas Farmacocinética clínica básica Ediciones Díaz de Santos, 1994 pgs. 8–14 ISBN 84-7978-147-5, 9788479781477 (in Spanish)
- Factors Affecting the Bioavailability of Chemicals Mikko Nikinmaa, in An Introduction to Aquatic Toxicology, 2014
- Di, Li; Kerns, Edward H. . Current Opinion in Chemical Biology. 2003-06-01, 7 (3): 402–408 [2023-05-08]. ISSN 1367-5931. doi:10.1016/S1367-5931(03)00055-3 (英语).
- Tetko IV, Bruneau P, Mewes HW, Rohrer DC, Poda GI. (PDF). Drug Discovery Today. August 2006, 11 (15–16): 700–707 [2023-05-02]. PMID 16846797. doi:10.1016/j.drudis.2006.06.013. (原始内容 (pre-print)存档于2013-09-12).
- Leeson, Paul D.; Springthorpe, Brian. . Nature Reviews Drug Discovery. 2007-11, 6 (11): 881–890 [2023-05-08]. ISSN 1474-1784. doi:10.1038/nrd2445. (原始内容存档于2023-01-28) (英语).
- Baaske P, Wienken CJ, Reineck P, Duhr S, Braun D. . Angewandte Chemie. March 2010, 49 (12): 2238–41. PMID 20186894. doi:10.1002/anie.200903998.
- Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. . Nature Communications. October 2010, 1 (7): 100. Bibcode:2010NatCo...1..100W. PMID 20981028. doi:10.1038/ncomms1093 .
- Hsieh Y, Korfmacher WA. . Current Drug Metabolism. June 2006, 7 (5): 479–89. PMID 16787157. S2CID 13612670. doi:10.2174/138920006777697963.
- Covey TR, Lee ED, Henion JD. . Analytical Chemistry. October 1986, 58 (12): 2453–60. PMID 3789400. doi:10.1021/ac00125a022.
- Covey TR, Crowther JB, Dewey EA, Henion JD. . Analytical Chemistry. February 1985, 57 (2): 474–81. PMID 3977076. doi:10.1021/ac50001a036.
- Committee for Medicinal Products for Human Use (CHMP). (PDF). European Medicines Agency, Evaluation of Medicines for Human Use. December 2009 [4 May 2013]. EMA/CPMP/ICH/286/1995. (原始内容存档 (PDF)于2018-06-15).
- Li, Xue; Martinez-Lozano Sinues, Pablo; Dallmann, Robert; Bregy, Lukas; Hollmén, Maija; Proulx, Steven; Brown, Steven A.; Detmar, Michael; Kohler, Malcolm; Zenobi, Renato. . Angewandte Chemie International Edition. 2015-06-26, 54 (27): 7815–7818. PMID 26015026. doi:10.1002/anie.201503312. hdl:20.500.11850/102558 (英语).
- Sheiner LB, Rosenberg B, Marathe VV. . Journal of Pharmacokinetics and Biopharmaceutics. October 1977, 5 (5): 445–79. PMID 925881. S2CID 28622472. doi:10.1007/BF01061728.
- Sheiner LB, Beal S, Rosenberg B, Marathe VV. . Clinical Pharmacology and Therapeutics. September 1979, 26 (3): 294–305. PMID 466923. S2CID 41194071. doi:10.1002/cpt1979263294.
- Bonate PL. . The AAPS Journal. October 2005, 7 (2): E363–73. PMC 2750974 . PMID 16353916. doi:10.1208/aapsj070237.
- Singh SS. . Current Drug Metabolism. February 2006, 7 (2): 165–182. PMID 16472106. doi:10.2174/138920006775541552.
- O'Valle, F.; García del Moral, R.; Andujar, M. . Nefrologia. 1995,. 15 Supplement 1 [2023-05-10]. (原始内容存档于2018-07-26) (西班牙语).
- Naesens, Maarten; Kuypers, Dirk R. J.; Sarwal, Minnie. . Clinical Journal of the American Society of Nephrology. 2009-02, 4 (2): 481 [2023-05-13]. ISSN 1555-9041. doi:10.2215/CJN.04800908. (原始内容存档于2023-07-10).
- Robert N, Wong GW, Wright JM. . Cochrane Database of Systematic Reviews. January 2010, (1): CD007893. PMID 20091657. doi:10.1002/14651858.CD007893.pub2.
- Rowland M, Tozer T (2010) Clinical pharmacokinetics and pharmacodynamics: concepts and applications, 4th edn. Lippincott Williams & Wilkins, Maryland
- Joaquín Herrera Carranza Manual de farmacia clínica y Atención Farmacéutica (in Spanish).
- Jager T, Albert C, Preuss TG, Ashauer R. . Environmental Science & Technology. April 2011, 45 (7): 2529–40. Bibcode:2011EnST...45.2529J. PMID 21366215. doi:10.1021/es103092a.
- US EPA, OMS. . www.epa.gov. 2016-11-17 [2022-05-15]. (原始内容存档于2023-09-22) (英语).
- . www.who.int. [2022-05-15]. (原始内容存档于2023-05-11) (英语).
- Ashauer R. . Swiss Federal Institute of Aquatic Science and Technology. [2011-12-03]. (原始内容存档于2012-04-05).
软件
- 非隔室模型
- 免费软件: R的bear (页面存档备份,存于)和PK (页面存档备份,存于) , Julia的MetidaNCA (页面存档备份,存于)
- 商业: MLAB 、 EquivTest (页面存档备份,存于) 、 Kinetica (页面存档备份,存于) 、 MATLAB/SimBiology (页面存档备份,存于) 、 PKMP (页面存档备份,存于) 、 Phoenix/WinNonlin (页面存档备份,存于) 、 PK Solutions 、 RapidNCA 。
- 基于膈室模型
- 免费软件: ADAPT (页面存档备份,存于) 、 Boomer (页面存档备份,存于) ( GUI (页面存档备份,存于) )、 SBPKPD.org(系统生物学驱动的药代动力学和药效学) (页面存档备份,存于) 、 WinSAAM 、 PKfit (页面存档备份,存于) for R、 PharmaCalc 和 PharmaCalcCL (页面存档备份,存于) 、Java 应用程序。
- 商业: PrecisePK (页面存档备份,存于) 、 Imalytics 、Kinetica、 MATLAB/SimBiology (页面存档备份,存于) 、 Phoenix/WinNonlin (页面存档备份,存于) 、PK Solutions、 PottersWheel 、 ProcessDB (页面存档备份,存于) 、 SAAM II (页面存档备份,存于) 。
- 基于生理学模型
- 免费软件: MCSim (页面存档备份,存于)、 PK-Sim (页面存档备份,存于)
- 商业: acslX 、 Cloe PK (页面存档备份,存于) 、 GastroPlus (页面存档备份,存于) 、 MATLAB/SimBiology (页面存档备份,存于) 、 ProcessDB (页面存档备份,存于) 、 Simcyp (页面存档备份,存于) 、 Entelos PhysioLab Phoenix/WinNonlin (页面存档备份,存于) 、 ADME Workbench 。
- 种群PK
- 免费软件: WinBUGS 、ADAPT、S-ADAPT / SADAPT-TRAN、Boomer、 PKBugs 、R 的Pmetrics (页面存档备份,存于) 。
- 商业: PrecisePK (页面存档备份,存于) 、Kinetica、 MATLAB/SimBiology (页面存档备份,存于) 、 Monolix (页面存档备份,存于) 、 NONMEM 、 Phoenix/NLME (页面存档备份,存于) 、SAAM II 的PopKinetics (页面存档备份,存于) 、 USC*PACK (页面存档备份,存于) 、 DoseMe-Rx (页面存档备份,存于) 、 Navigator Workbench 。
- 治疗药物监测(TDM)
- 模拟
以上所有基于模型的软件。
教育中心
在提供深度培训方面知名度最高的全球中心包括布法罗大学药学和药物科学学院、佛罗里达大学药学院药剂学及临床药物科学系、北卡罗来纳大学药学院药物实践与实验疗法系、明尼苏达大学双城分校药学院药剂系及实验与临床药物科学系、乌普萨拉大学、哥德堡大学、莱顿大学、奥塔哥大学、旧金山大学、北京大学附属第三医院、东京大学、华盛顿大学药学院药剂系、曼彻斯特大学、莫纳什大学、谢菲尔德大学和复旦大学药学院临床药学系。 [1]
- Tucker GT. . British Journal of Clinical Pharmacology. June 2012, 73 (6): 924–6. PMC 3391520 . PMID 22360418. doi:10.1111/j.1365-2125.2012.04238.x.