| 2-Hydroxyglutaric aciduria | |
|---|---|
 | |
| Alpha-Hydroxyglutaric acid |
2-hydroxyglutaric aciduria is a rare neurometabolic disorder characterized by the significantly elevated levels of hydroxyglutaric acid in one's urine. It is either autosomal recessive or autosomal dominant.[1]
Presentation
The signs/symptoms of this condition are consistent with the following:[2]
Cause
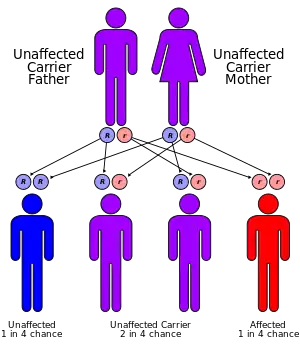
Mutation in several genes can lead to different types of 2-hydroxyglutaric aciduria. For example, the D2HGDH and L2HGDH genes provide instructions for making enzymes that are found in mitochondria - in which these enzymes break down D-2-hydroxyglutarate and L-2-hydroxyglutarate, respectively, as a part of normal reaction series that generate energy for cell activities. Any mutations occur in either of these genes would interrupt the functional enzymes and allow both 2-hydroxyglutarates to accumulate in cells, which cause 2-hydroxyglutaric aciduria type I. Moreover, it is known that type II for L-2-hydroxyglutaric aciduria and a mixed type for both 2-hydroxyglutarates come from mutations in IDH2 gene and SLC25A1 gene, respectively.[3][4]
Diagnosis
Classification
2-hydroxyglutaric aciduria is an organic aciduria, and because of the stereoisomeric property of 2-hydroxyglutarate different variants of this disorder are distinguished:
L-2-hydroxyglutaric aciduria
The L-2 form is more common, severe, and mainly affects the central nervous system. The basal ganglia are affected, and cystic cavitations in the white matter of the brain are common, beginning in infancy. This form is chronic, with early symptoms such as hypotonia, tremors, and epilepsy declining into spongiform leukoencephalopathy, muscular choreodystonia, mental retardation, and psychomotor regression.[5]
It is associated with L2HGDH, which encodes L-2-hydroxyglutarate dehydrogenase.[6] L-2-hydroxyglutarate is produced by promiscuous action of malate dehydrogenase on 2-oxoglutarate, and L-2-hydroxyglutarate dehydrogenase is an example of a metabolite repair enzyme that oxidizes L-2-hydroxyglutarate back to 2-oxoglutarate.[7]
D-2-hydroxyglutaric aciduria
The D2 form is rare, with symptoms including macrocephaly, cardiomyopathy, mental retardation, hypotonia, and cortical blindness.[8] It is caused by recessive mutations in D2HGDH[9] (type I) or by dominant gain-of-function mutations in IDH2[10] (type II).
Combined D-2- and L-2-hydroxyglutaric aciduria
The combined form is characterized by severe early-onset epileptic encephalopathy and absence of developmental progress.[11] It is caused by recessive mutations in SLC25A1 encoding the mitochondrial citrate carrier.[12]
Treatment
The treatment of 2-Hydroxyglutaric aciduria is based on seizure control, the prognosis depends on how severe the condition is.[13]
See also
References
- ↑ Reference, Genetics Home. "2-hydroxyglutaric aciduria". Genetics Home Reference. Retrieved 25 January 2017.
- ↑ "L-2-hydroxyglutaric aciduria | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 25 January 2017.
- ↑ "2-hydroxyglutaric aciduria: MedlinePlus Genetics". medlineplus.gov. Retrieved 2022-07-17.
- ↑ Kranendijk, Martijn; Struys, Eduard A.; Salomons, Gajja S.; Van der Knaap, Marjo S.; Jakobs, Cornelis (2012). "Progress in understanding 2-hydroxyglutaric acidurias". Journal of Inherited Metabolic Disease. 35 (4): 571–587. doi:10.1007/s10545-012-9462-5. ISSN 0141-8955. PMC 3388262. PMID 22391998.
- ↑ Seijo-Martinez M, Navarro C, Castro del Rio M, Vila O, Puig M, Ribes A, Butron M (2005). "L-2-hydroxyglutaric aciduria: clinical, neuroimaging, and neuropathological findings". Arch. Neurol. 62 (4): 666–670. doi:10.1001/archneur.62.4.666. PMID 15824270.
- ↑ Topçu M, Jobard F, Halliez S, et al. (November 2004). "L-2-Hydroxyglutaric aciduria: identification of a mutant gene C14orf160, localized on chromosome 14q22.1". Hum. Mol. Genet. 13 (22): 2803–11. doi:10.1093/hmg/ddh300. PMID 15385440.
- ↑ Van Schaftingen, E.; Rzem, R.; Veiga-da-Cunha, M. (2009-04-01). "L: -2-Hydroxyglutaric aciduria, a disorder of metabolite repair". Journal of Inherited Metabolic Disease. 32 (2): 135–142. doi:10.1007/s10545-008-1042-3. ISSN 1573-2665. PMID 19020988. S2CID 27702186.
- ↑ Nyhan WL, Shelton GD, Jakobs C, Holmes B, Bowe C, Curry CJ, Vance C, Duran M, Sweetman L (1995). "D-2-hydroxyglutaric aciduria". J. Child Neurol. 10 (2): 137–142. doi:10.1177/088307389501000216. PMID 7782605. S2CID 19395651.
- ↑ Struys EA, Salomons GS, Achouri Y, Van Schaftingen E, Grosso S, Craigen WJ, Verhoeven NM, Jakobs C (Jan 2005). "Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria". Am J Hum Genet. 76 (2): 358–60. doi:10.1086/427890. PMC 1196381. PMID 15609246.
- ↑ Kranendijk M, Struys EA, van Schaftingen E, et al. (2010). "IDH2 mutations in patients with D-2-hydroxyglutaric aciduria". Science. 330 (6002): 336. Bibcode:2010Sci...330..336K. doi:10.1126/science.1192632. PMID 20847235. S2CID 206527781.
- ↑ Muntau A, Röschinger W, Merkenschlager A, van der Knaap MS, et al. (2000). "Combined D-2- and L-2-hydroxyglutaric aciduria with neonatal onset encephalopathy: a third biochemical variant of 2-hydroxyglutaric aciduria?". Neuropediatrics. 31 (3): 137–40. doi:10.1055/s-2000-7497. PMID 10963100.
- ↑ Nota B; et al. (2013). "Deficiency in SLC25A1, Encoding the Mitochondrial Citrate Carrier, Causes Combined D-2- and L-2-Hydroxyglutaric Aciduria". The American Journal of Human Genetics. 92 (4): 627–631. doi:10.1016/j.ajhg.2013.03.009. PMC 3617390. PMID 23561848.
- ↑ "D-2-hydroxyglutaric aciduria". OrphaNet: The portal for rare diseases and orphan drugs.