| MPC1 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | MPC1, BRP44L, MPYCD, dJ68L15.3, CGI-129, mitochondrial pyruvate carrier 1, SLC54A1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 614738 MGI: 1915240 HomoloGene: 9384 GeneCards: MPC1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||


Mitochondrial pyruvate carrier 1 (MPC1), also known as brain protein 44-like (BRP44L) and SLC54A1, is a protein that in humans is encoded by the MPC1 gene.[5] It is part of the Mitochondrial Pyruvate Carrier (MPC) protein family. This protein is involved in transport of pyruvate across the inner membrane of mitochondria in preparation for the pyruvate dehydrogenase reaction.
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]
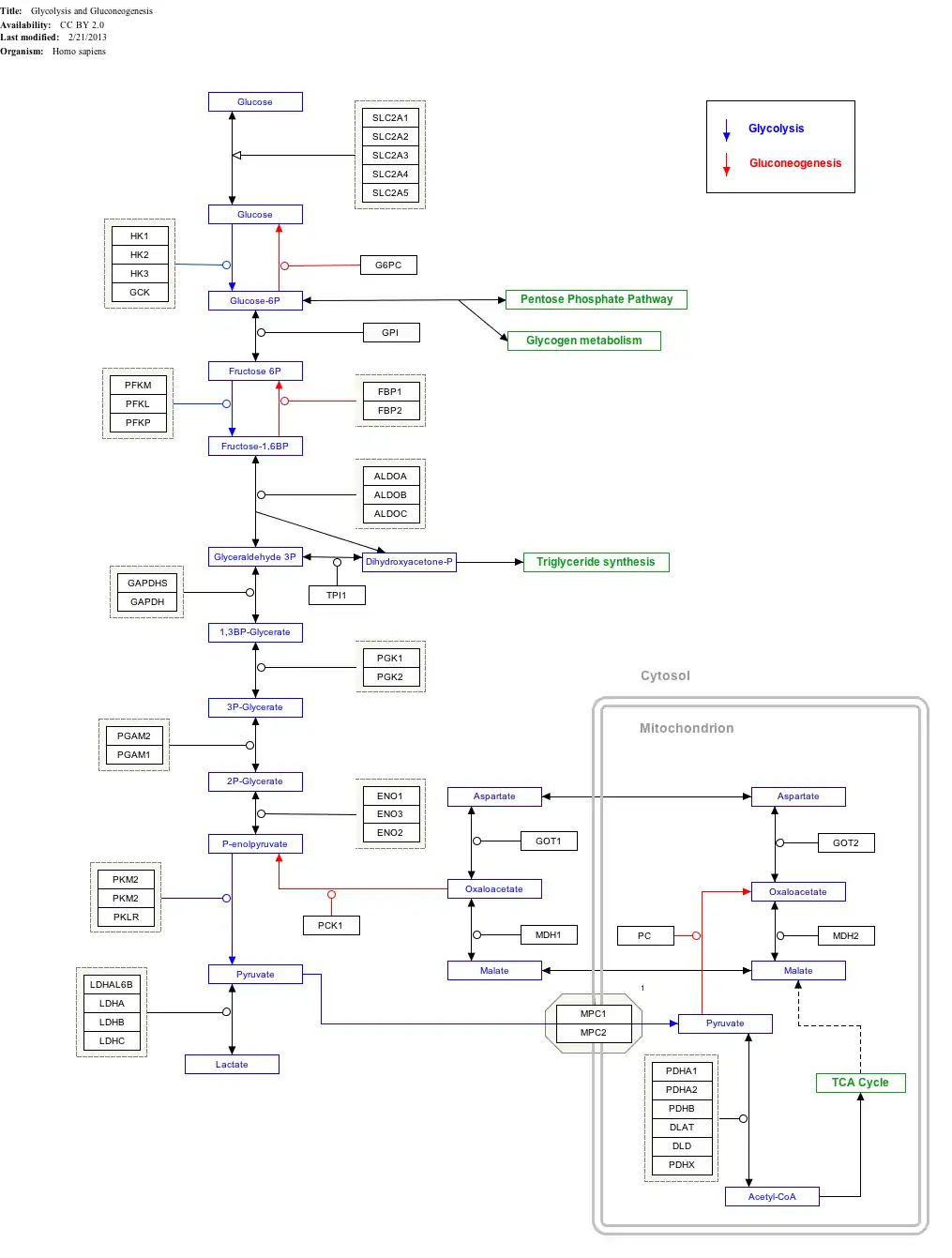
- ↑ The interactive pathway map can be edited at WikiPathways: "GlycolysisGluconeogenesis_WP534".
Clinical significance
Mitochondrial pyruvate carrier deficiency (MPYCD) is an autosomal recessive disease due to mutations in the MPC1 gene on chromosome 6q27. It is an inborn error of carbohydrate metabolism that blocks aerobic glycolysis by preventing the transport of pyruvate from the cytosol into the mitochondrion for oxidative phosphorylation; however, anaerobic glycolysis is preserved. Common signs and symptoms include poor growth, normal lactate/pyruvate ratio (however both lactate and pyruvate are in higher than normal concentrations), hepatomegaly, lactic acidosis, hypoglycemia, neurological problems, and hypotonia.[6] A disease with comparable symptoms is also seen in autosomal recessive mutations of the MPC2 gene.[7]
See also
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000060762 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000023861 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "HGNC data for MPC1". HUGO Gene Nomenclature Committee. Retrieved 2023-08-21.
- ↑ "Mitochondrial pyruvate carrier 1; MPC1". Online Mendelian Inheritance in Man (OMIM). Retrieved 2023-08-21.
- ↑ Pujol C, Lebigot E, Gaignard P, Galai S, Kraoua I, Bault JP, et al. (March 2023). "MPC2 variants disrupt mitochondrial pyruvate metabolism and cause an early-onset mitochondriopathy". Brain. 146 (3): 858–864. doi:10.1093/brain/awac444. PMC 9976959. PMID 36417180.