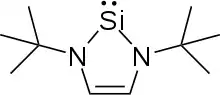
An N-Heterocyclic silylene (NHSi) is an uncharged heterocyclic chemical compound consisting of a divalent silicon atom bonded to two nitrogen atoms. The isolation of the first stable NHSi, also the first stable dicoordinate silicon compound, was reported in 1994 by Michael Denk and Robert West three years after Anthony Arduengo first isolated an N-heterocyclic carbene, the lighter congener of NHSis.[1][2] Since their first isolation, NHSis have been synthesized and studied with both saturated and unsaturated central rings ranging in size from 4 to 6 atoms. The stability of NHSis, especially 6π aromatic unsaturated five-membered examples, make them useful systems to study the structure and reactivity of silylenes and low-valent main group elements in general. Though not used outside of academic settings, complexes containing NHSis are known to be competent catalysts for industrially important reactions. This article focuses on the properties and reactivity of five-membered NHSis.
Synthesis and stability
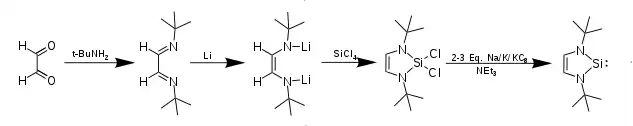
Five-membered N-heterocyclic silylenes are synthesized from two-carbon diimine or diamine precursors. Reaction with metallic lithium yields a lithiated diimide or diamine that when stirred with tetravalent SiCl4 yields a tetravalent dichlorosilicocycle. The compound then is reduced with an alkali metal, or potassium graphite to reveal a divalent silicon center. Care must be taken to not over reduce the silicon; often triethylamine is added to prevent over reduction. The yield for this reduction can be as high as 80%.[3][4] N-heterocyclic silylenes containing Silicon(II) with four-membered rings (1 carbon) and six-membered rings (3 carbons) are well known and derived from the reactions of SiX4 with amidinate and NacNac ligands respectively.[4]
The silylenes synthesized prior to West and Denk's first NHSi decomposed at temperatures below 77K. Alternatively, the original West/Denk NHSi [tBuN−CH=CH−tBuN]Si: is exceptionally stable even after heating for 4 months at 150 °C dissolved in toluene in a sealed NMR tube. It is also unreactive with Lewis bases and triethylsilane a silylene scavengers, but it does react with air and water (see below) and decomposes at its melting point 220 °C.[1][3] On the other hand, the saturated NHSi with no backbone substitutions, [tBsN−CH2CH2−tBuN]Si:, is far more reactive as a Lewis acid and far less stable, decomposing at 25 °C.[2]
Structure and aromaticity

Structure
Five-membered NHSis can be classified by the bonding in the two-carbon backbone of their rings. Saturated NHSis have two methylene units, the unsaturated have doubly-bonded methine carbons, and the benzo-fused NHSis share their carbon backbone with a fused aromatic ring. Though other N substituents such as aryl groups are also well known, in general, studies are conducted with tert-butyl groups bonded the nitrogen of the saturated and unsaturated NHSis, while the benzo-fused have neopentyl groups.
X-ray crystallography and electron diffraction show significant differences between the saturated and unsaturated structures. The N-Si-N angle for the saturated tert-butyl substituted NHSi is 92°, slightly puckering C2 symmetric ring. The N-Si-N angle of the unsaturated analog is 90.5° (gas phase XRD) achieving a planar C2v ring. Upon saturation of the carbon backbone, the N-Si bonds shorten from 175.3pm to 171.9pm, shorter distances than would be expected for a single bond between nitrogen and divalent silicon. The longer bond in the unsaturated molecule is due to the nitrogens' lone pair electrons delocalization through the carbon π-bond, resulting in lower Lewis basicity of the nitrogen lone pairs toward the silicon, weakening and lengthening the N-Si bond. The overall silylene character is supported by strong downfield shifting in 29Si NMR at +78.3ppm for the unsaturated at +119ppm for the NHSis compared to their tetravalent dichloride starting materials which resonate at -40.7ppm.[2][5]
Aromaticity
The structural differences, distinct reactivity, and increased stability of the unsaturated ring is attributed the olefinic C=C bond which creates a 6π aromatic ring with the two nitrogen lone pairs. The Si-N bond length discrepancy mentioned above is a strong indication of π delocalization. In proton NMR the vinylic protons resonate at 6.75ppm for [tBuN−CH=CH−tBuN]Si: versus 5.73ppm for the tetravalent dichloride starting material or 6.00ppm for the tetravalent NHSi-H2 dihydride. In the tetravalent species, the lack of the silicon's empty 3p orbital breaks the π ring, causing an upfield shift as the deshielding due to aromatic ring current is lost.[2] Additionally there are significant discrepancies in the Raman spectroscopy signals between the unsaturated NHSi and tetravalent silicon analogues, such as NHSi-Cl2 and spiro-[tBuN−CH=CH−tBuN]2Si. The four-coordinate silanes exhibit C=C stretches at 1620 cm−1. Conversely [tBuN−CH=CH−tBuN]Si:'s C=C resonance is found bifurcated at 1566 cm−1 and 1573 cm−1 at a much greater intensity. In the divalent species, electron density is shifted out of the C=C bond into the ring and empty Si 3p orbital, weakening the C=C bond and causing the observed energy drop and shape change in the C=C resonance.[6]
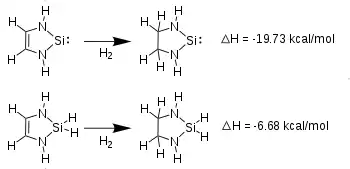
Several computational studies suggest aromatic delocalization over the unsaturated NHSi rings. DFT calculations of NHSi-phosphonyl radical adducts show far greater radical electron delocalization over the rings of unsaturated NHSis (14% Si, 65% ring) than the saturated NHSi (58% Si, 19% ring).[7] Aromaticity is further supported by calculated heats of hydrogenation. At the MP4/6-31G*//6-31G* level, hydrogenation at the carbon backbone of an unsaturated divalent silicon NHSi is 13 kcal/mole more exothermic than the corresponding hydrogenation of the tetracoordinate-silane analog. The stabilization of the divalent silicon ring, with an empty 3p orbital and in-ring lone pair, versus the tetravalent silane which lacks an empty Si 3p orbital indicates significant electronic communication between the silicon and the olefinic carbons.[5] Additionally, calculations of Nuclear Independent Chemical Shift of the ring-centered NMR resonances of [HN-CH=CH-NH]Si: at -10.2ppm indicate a high degree of delocalization about the unsaturated NHSi ring. In reference to the perfectly aromatic cyclopentadienyl anion's shift at -14.3ppm, NICS data supports the aromaticity of NHSis.[8]
Electronics
Computational studies and photoelectron spectroscopy create a more nuanced picture of electronic structure and π-electron sharing in NHSis. Relevant π bonding orbitals in unsaturated five-membered NHSis can be understood as the molecular orbital mixing of the diimine backbone's butadiene-like pi orbitals and the 3s and 3p orbitals of Si. Photoelectron Spectra of the unsaturated [tBuN−CH=CH−tBuN]Si: show a HOMO with three peaks at -6.96eV, -7.13eV & -7.28eV, assigned the in-phase overlap of the Si 3py orbital and buta-1,3-diene-like π3 of the diimine. The HOMO in the tetravalent Si-hydrogenated analog lacks a vacant Si 3py orbital and the energy of the π3 is destabilized to -6.56 eV, supporting charge donation from N to Si in the silylene. The HOMO-1 is the in-plane Si lone pair involved in σ donation. No difference is calculated between the HOMO-2 for the unsaturated silylene compared to the corresponding orbital of the silane analog. The original 1994 Denk at al. photoelectron spectroscopy communication relates this orbital to the π2 diimine orbital, however a 1996 paper from Blakeman et al. designates this as the nitrogen lone pairs. Either way, both orbital assignments are symmetrically forbidden from overlapping with any Si orbital. The HOMO-2 does not change in energy upon hydrogenation at Si indicating that much of the overall electronics are unchanged by hydrogenation and underscoring the effects of N to Si electron π-donation in the HOMO and HOMO-3. Calculations in the former study at the MP4 6-31G* level showed increased energy level spacing for N-H model compounds (instead of N-tBu), but with the same order and proportional spacing.[5][9] The Blakeman study compared the benzo-fused NHSi to the ethylene-like unsaturated NHSi, and found by PES that the HOMO is lower by .36eV and the Si lone pair by .34 eV in the benzo-fused NHSi, suggesting the electron withdrawing power of the phenylene ring makes the benzo-fused NHSi less nucleophilic.[9]
| Saturated NHSi (C2) | Unsaturated NHSi (C2V) | NHSilane (C2V) |
|---|---|---|
| LUMO | LUMO | LUMO |
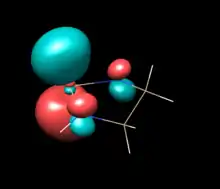 |
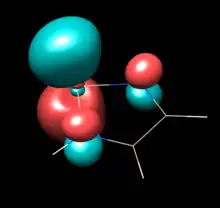 |
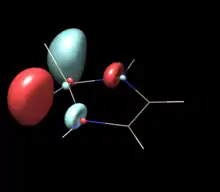 |
| HOMO (B) | HOMO (B2) | HOMO (B2) |
| -7.54 eV (calc. -8.76) | -6.94, -7.13, -7.28 eV (calc. -7.62) | -6.56 eV (calc. -7.10) |
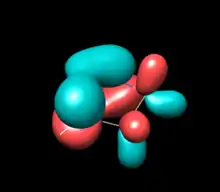 |
 |
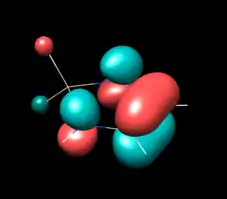 |
| HOMO - 1 (A) | HOMO - 1 (A1) | |
| -8.11 eV (calc. -9.40) | -8.21 eV (calc. -9.07) | |
 |
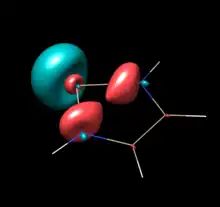 |
No corresponding orbital
No empty Si 3pπ |
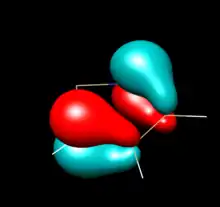
| HOMO - 2 (A2) | HOMO -1(A2) | |
| -9.08 eV (calc. -11.32) | -8.92 eV (calc. -11.32) | |
| No corresponding orbital
There are no p orbitals on saturated carbons to mix with nitrogen lone pairs |
 |
 |
The above table show orbitals, labeled above by occupation and grouped horizontally based on symmetry orbital parentage. They are reproductions of calculations performed in Denk, Green et al.1994 and Blakeman et al.. Experimental energies are from PES of original West/Denk-like tert-butyl NHSis, calculated energies are from N,N' hydride-like NHSi shown in MO graphic. (LUMO energies and symmetries were not provided.)
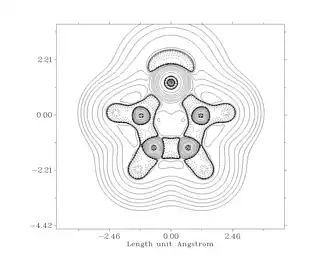
Natural Bond Orbital analysis of [HN-CH=CH-NH]Si: provides strong theoretical evidence of π electron delocalization. NBO calculations indicate a population of 0.54 electrons in the formally vacant Si 3py orbital (1.2 electrons would be expected in a pure 6 π-electron 5 atom ring). The saturated silylene has lower electron delocalization over the heteroallylic N-Si-N system with only 0.33 electrons on Si (versus 1.33 for perfect 4 electron shared among 3 atoms). No difference in charge sharing is seen in the σ-bond interaction between Si and N of each NHSi, this is also the case with NHCs. In spite of identical Si-N σ bonding, the unsaturated silylene has a lower charge separation in its Si-N bond than the saturated analog: for the unsaturated analog, Si has a charge of +0.87 and each N -1.00, while the saturated has a charge of +1.05 on Si and -1.12 on N. These data show that the stabilization and bond difference between the backbone types is entirely due to π interactions. For comparison, in an analogous way, germylenes have a charge near +1 at the germanium while cabenes have close to no net charge on the carbenic carbon. Calculations show, upon coordination to Group 11 metal chlorides (e.g. NHSi-CuCl) Si 3py density, N-Si bond density, and Si atom charge all increase. The change is the result of Si's sigma donation to the metal, which causes the Si 3p orbital to draw electron density from the ring. In this manuscript they calculate CuCl and AgCl back-donation to Si to be about 4.5 weaker than the σ interaction. Interestingly, for all three Group 11 metals, the NHSi in question is a better σ donor and π acceptor than its germylene and carbene analogs.[10][8]
Atoms in Molecules bond energy at critical point calculations show that the N to Si bonds in an NHSi are significantly ionic, unlike the highly covalent N-C bond in an NHC. Furthermore, the charge density and the Laplacian (∇2) at the bond critical points between the silylene and carbene are dissimilar. Silylenes have much lower charge density between Si and N, with a positive Laplacian from Si to N indicating a more ionic interaction, while the Laplacian is negative going from carbon to nitrogen in an NHC indicating a more covalent bond. Upon coordination to a Group 11 metal the covalent character in the Si or C to metal bond is small, but not insignificant, again with a higher covalent character in the carbenic system. Similarly, silicon has a higher Laplacian between it and the group 11 metals than does carbon, indicating a less covalent metal interaction in silyenes as compared to carbenes.[8]
Main group reactivity
N-heterocyclic silylenes react with a wide variety of main group compounds, generally adding across unsaturated bonds or inserting into single bonds to yield tetravalent N-heterocyclic silanes.
Tetramerization
In concentrated solutions, the saturated unsubstituted silylene [tBuN−CH2CH2−tBuN]Si: reacts with itself to form a tetramer with a Si-Si double bond. First one NHSi inserts its divalent silicon into another's N-Si bond, creating a bicyclic molecule with a tetravalent silicon bonded to a divalent silylene. The mixed-valent dimer then itself dimerizes, forming a four-silicon silene with 229 pm Si=Si double bond. Though exceptionally long, the bond order is supported by Raman spectroscopy. Reacting the tetramer with methanol results in the mixed valent-dimer-methanol adduct and not the silene-dimer adduct (see below). Additionally a double bond between tert-butyl substituted NHSis is calculated to not be stable, further corroborating the identity of the mixed valent dimer as the intermediate.[11] Unsaturated and backbone-substituted saturated NHSis such as the 4,5-dimethyl-[tBuN−CHMeCH−tBuN]Si: are not reactive in this way and as remain monomers.[12] Similarly, the unsubstituted NHSis will react with analogous N heterocyclic germylenes, ultimately forming a silicon-flanked germene.[3]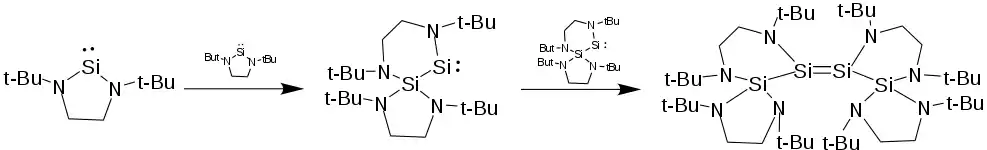
Reactions with chalcogens
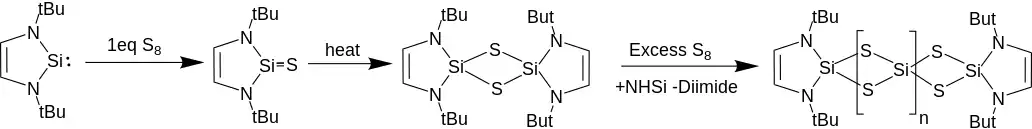
Reacting NHSis with oxygen strips the diimine backbone, and produces solid silicon dioxide, likely through a two NHSi dioxo-bridged intermediate. Reactions with 1 equivalent of S8 yields a dithio-bridged two NHSi complex. The formation of the dithio dimer occurs through an intermediate attributed to a thiosilylone, with a Si-S double bond. Excess sulfur yields a repeating NHSi(S2)(SiS2)nNHSi oligomer with complimentary diimine loss. The analogous reaction with selenium is far slower, but yields the diselenium bridged di-NHSi exclusively.[13]
Reactions with σ bonds
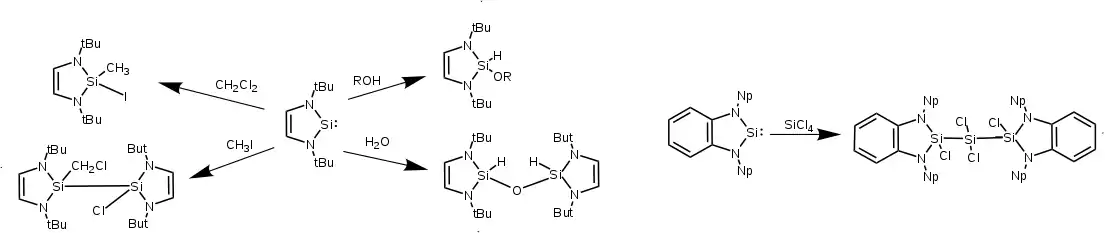
NHSis insert into σ bonds to form terminal silane-like products, NHSi-NHSi disilane bridges, or oxo-bridged NHSi siloxanes. Alcohols react with Si(II) to form exclusively the silyl ether. With water, two NHSis will insert to each of the O-H bonds to form a siloxane. A more varied reactivity is seen with haloalkanes. NHSis insert directly into the C-I bond of iodomethane. Alternatively, when saturated or unsaturated NHSis are reacted with dichloromethane, chloroform, or carbon tetrachloride, the silicon centers of two NHSis form a bond, with one halide adding to one silicon and the remaining organic fragment adding to the other. Other halocarbons will yield a mixture of mono and disilane products. The resulting mixture of products as well as computational studies indicate that a free radical mechanism brings about these products, with the radical pathway being 16 to 23 kcal/mol lower in energy than concerted mechanisms. Upon heating, the disilanes either disproportionate to an NHSilane and NHSilylene, or form a one-carbon bridge between two NHSi-X fragments in a vein similar to water's reactivity. In the presence of silicon tetrachloride, benzo-fused NHSis will insert between two Si-Cl bonds to form a trisilane.[14][4]
Reactions with π bonds
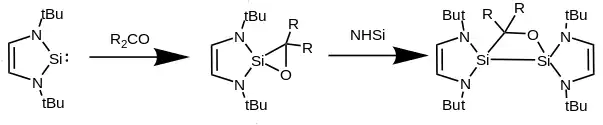
Generally, when reacting with unsaturated species an NHSi will add across a double or triple bond to form a spirocyclic compound, containing one or two equivalents of NHSi-derived silicon. Alkenes and alkynes react with NHSis only if their C-C bond is highly polarized such as is the case with phenyltrimethylsilylacetylene, which produces a silicocyclopropene with one NHSi equivalent or a disilicocyclobutene with two. With ketones two equivalents of benzo-fused NHSi react to form disilyloxetanes as well as further reaction products depending on the carbon substituents.
Dienes, as well as heterodienes including benzil or the common NHSi backbone N,N'-diterbutyl-1,4diazabutadiene ([tBuN-CH=CH-tBuN]) react in a [4+1] fashion to yield molecules with two five-membered rings sharing a single silicon atom.[2][4]

Imines do not form simple cyclic silylene additions: benzo-fused NHSis react with the diimine and ortho carbons of N-tert-butyl phenylimine to form a five-membered ring composed of the NHSi silicon, one carbon and nitrogen of the imine and the ipso and para carbons of the phenyl group, through an intermediate that breaks the phenyl aromaticity.[15]

NHSis add across nitrile C-N bonds, first forming a three-membered ring, subsequently a second NHSi inserts to complete a 4-membered ring, similar to the reactivity of a alkyne. Alternatively, tert-butyl isocyanide reacts more like an alkyl halide with a single silicon bonding to the tert-butyl fragment and the carbon of the cyanide groups, or with two NHSis to from an acyclic tBu-Si-Si-CN product.[16] There is evidence that the single-silicon NHSi-nitrile interconverts in small quantities to the silicon isonitrile.[3]
As a Lewis acid
N-heterocyclic silylenes are generally considered to be Lewis bases, though their reactivity can occasionally differ. With the Lewis acid tris(pentafluorophenyl)borane, the expected acid/base adduct forms, however, over long periods of time, the silicon will insert between a B-C bond, oxidizing the silyene to a silane.[17] Additionally, in rare cases, an NHSi can act as a Lewis acid. Reaction of the N,N' dineopentyl beznofused NHSi with the analogous carbene, creates a long, highly polarized C-Si bond. The carbene carbon becomes a carbocation, remaining nearly planar, while the pyramidalized silicon adopts a negative charge.[18][3]
Transition metal coordination
N-Heterocyclic silylenes as ligands
Though with less variety and fewer applications than their lighter analogues, the N-heterocyclic carbenes, N-heterocyclic silylenes are known to act as ligands in a range of metal complexes. Unlike most non-NHSi silylenes which are Lewis acidic, stable five-membered NHSis act almost exclusively as a Lewis bases or nucleophiles in a manner similar to NHCs and phosphine ligands.[2] Both the saturated and unsaturated five-membered NHSis are calculated to have substantial singlet-triplet energy gaps, at 74 kcal/mol and 69 kcal/mol respectively for the hydride N-substituted models.[5] As a result of the large energy gap, electron density is concentrated exclusively in the in-plane σ-donating Si lone pair while π donation from the ring stabilizes the formally vacant, slightly π-accepting Si pπ orbital.[2] Though weak, as discussed above, NHSis do exhibit metal to Si 3p π-backbonding, as evidenced by the shortness of NHSi-metal bonds compared to standard metal-Si single bonds. In the complex [tBuN−CH=CH−tBuN]Si:–Fe(CO)4, the NHSi readily takes the place of strongly backbonding carbonyls. For the trigonal-bipyramidal iron, the NHSi adopts an equatorial position over axial, with shorter equatorial Fe-Si distance (2.196Å vs 2.237Å) due to better overlap of the empty acidic 3p orbital with the metal d orbitals at this position.[19] Sterically, NHSis resemble NHCs, however, the tighter N-Si-N angle (c. 90°) and longer Si-N (c. 1.73Å) and Si-metal bond length compared to a carbene's N-C-N angle (c. 100°), C-N bond length (c. 1.37Å), and C-metal length means NHSi are less sterically demanding, leading to reactivity not seen in carbenes.[4]
Transition metal complexes and reactivity
N-heterocyclic silylenes readily substitute carbonyl, cyclooctadiene, and phosphine ligands in metal complexes. Additionally, silylenes can react with metals by inserting between metal ligand bonds or by acting as a reducing agent. [tBuN−CH=CH−tBuN]Si: can form two distinct molybdocene adducts depending on molybdenum precursor. With Mo(Cp)2(PEt3) the phosphine is replaced, while with Mo(Cp)2H2 an NHSi inserts between a metal-hydride bond to form a tetravalent silane ligand. The orbitals of the triethylphosphine substitution product are unable to form π interaction as exhibited by a 29Si NMR signal of 139.3ppm which indicates silicenium (Si+) character, while the molybdosilane has a resonance at 43.6ppm.[20][19]
As mentioned above, unsaturated NHSi reacts with Fe2(CO)9 to form both the axial and equatorial Fe(CO)4NHSi. Similarly, the same NHSi reacts with Ru3(CO)12 to form the axially trans-disubstituted Ru(CO)3(NHSi)2.[19] Reaction of [tBuN−CH=CH−tBuN]Si: with Cp*Ru(MeCN)3OTf creates Cp*RuCl(MeCN)2(NHSi)OTf in high yield. Upon heating, two equivalents of Cp*RuCl(MeCN)2(NHSi)+ lose a total of 2 acetonitrile ligands and one NHSi to form a diruthenium complex where one RuCp* unit is coordinated by the NHSi in a η1 fashion, while the other Cp*Ru center is complexed in a η5 fashion by the 6 electron putatively aromatic surface formed by the unsaturated NHSi ring.[21]
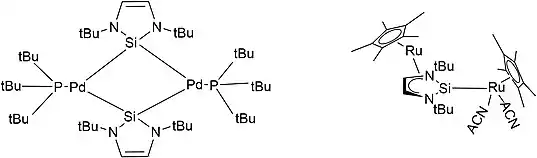
NiCl2(PPh3)2 reacts with 4 equivalents of benzo-fused NHSi to form Ni(NHSi)3(PPh3) while 5 equivalents are needed to produce the tetrakis NHSi complex. The extra NHSi equivalent is needed to reduce the Ni center, forming 1 equivalent of NHSiCl2 in the process. Both the saturated and unsaturated five-membered NHSis react with Pd(P(t−but)3)2, producing a two Pd complex with two terminal and two bridging NHSis. This unstable intermediate converts to a more stable complex with bridging silylenes and two terminal phosphines. Likewise, Pd(cod)2 starting materials are unable to produce Pd(NHSi)4, however, reduction of Pd(cod)Me2 with 6 equivalents of NHSi produces Pd[NHSiunsaturated)]3 or Pd[NHSiunsaturated)]4. Pd[NHSiunsaturated)]3 can also be produced by reacting Pd coordinated by just two N,N' tert-butyl NHCs with 3 equivalents of NHSi. Two factors contribute to this difference: the greater stearic bulk of the NHC makes Pd able to accommodate only 2 ligands and, calculations show, the NHSi has a 3.6 kcal/mol stronger Pd bond than the NHC.[22][4]
Beyond being interesting models to study the structure and bonding of low valent silicon, NHSis transition metal complexes have been demonstrated as active catalysts in industrially important reactions. The di-silylene bridged palladium phosphine complex, first synthesized in 2001, has palladium in the 0th oxidation state and is effective at Suzuki coupling. At 5% catalyst loading the easily synthesized NHSi bridged Pd complex is able to form a carbon carbon bond between phenylboronic acid and o-bromoacetophenone at 88% yield.[23] In another example, Pd(NHSi)(Cl)(C3H5), synthesized from [Pd(Cl)(C3H5)]2 and [tBuN−CH=CH−tBuN]Si: was found to be more effective than its NHC analogue at Heck coupling. With 1% catalyst loading at 140 °C the Pd(II) silylene complex catalyzes the coupling of p-bromoacetophenone and styrene in four hours in quantitative 99%+ yield. Under the same conditions the corresponding NHC complex yields only 91% product, showing that there are instances where the electronics or stearics do make silylenes comparatively superior catalysts.[24]
References
- 1 2 Denk, Michael; Lennon, Robert; Hayashi, Randy; West, Robert; Belyakov, Alexander V.; Verne, Hans P.; Haaland, Arne; Wagner, Matthias; Metzler, Nils (1994-03-01). "Synthesis and Structure of a Stable Silylene". Journal of the American Chemical Society. 116 (6): 2691–2692. doi:10.1021/ja00085a088. ISSN 0002-7863.
- 1 2 3 4 5 6 7 West, R.; Denk, Michael (1996-01-01). "Stable silylenes: synthesis, structure, reactions". Pure and Applied Chemistry. 68 (4): 785–788. doi:10.1351/pac199668040785. ISSN 1365-3075.
- 1 2 3 4 5 Haaf, Michael; Schmedake, Thomas A.; West, Robert (2000-10-01). "Stable Silylenes". Accounts of Chemical Research. 33 (10): 704–714. doi:10.1021/ar950192g. ISSN 0001-4842. PMID 11041835.
- 1 2 3 4 5 6 Asay, Matthew; Jones, Cameron; Driess, Matthias (2011-02-09). "N-Heterocyclic Carbene Analogues with Low-Valent Group 13 and Group 14 Elements: Syntheses, Structures, and Reactivities of a New Generation of Multitalented Ligands†". Chemical Reviews. 111 (2): 354–396. doi:10.1021/cr100216y. ISSN 0009-2665. PMID 21133370.
- 1 2 3 4 Denk, Michael; Green, Jennifer C.; Metzler, Nils; Wagner, Matthias (1994-01-01). "Electronic structure of a stable silylene: photoelectron spectra and theoretical calculations of Si(NRCHCHNR), Si(NRCH2CH2NR) and SiH2(NRCHCHNR)". Journal of the Chemical Society, Dalton Transactions (16): 2405. doi:10.1039/dt9940002405. ISSN 0300-9246.
- ↑ Leites, L.A; Bukalov, S.S; Denk, M; West, R; Haaf, M (2000-09-01). "Raman evidence of aromaticity of the thermally stable silylene ( t BuNCH CHN t Bu)Si". Journal of Molecular Structure. 550–551: 329–335. doi:10.1016/s0022-2860(00)00395-1. ISSN 0022-2860.
- ↑ Sheberla, Dennis; Tumanskii, Boris; Tomasik, Adam C.; Mitra, Amitabha; Hill, Nicholas J.; West, Robert; Apeloig, Yitzhak (2010-01-01). "Different electronic structure of phosphonyl radical adducts of N-heterocyclic carbenes, silylenes and germylenes: EPR spectroscopic study and DFT calculations". Chemical Science. 1 (2): 234. doi:10.1039/c0sc00143k. ISSN 2041-6520.
- 1 2 3 Boehme, Christian; Frenking, Gernot (1998-12-01). "N-Heterocyclic Carbene, Silylene, and Germylene Complexes of MCl (M = Cu, Ag, Au). A Theoretical Study1". Organometallics. 17 (26): 5801–5809. doi:10.1021/om980394r. ISSN 0276-7333.
- 1 2 Blakeman, Philip; Gehrhus, Barbara; Green, Jennifer C.; Heinicke, Joachim; Lappert, Michael F.; Kindermann, Markus; Veszprémi, Tamás (1996-01-01). "Electronic structure of stable benzodiazasilylenes: photoelectron spectra and quantum-chemical investigations". J. Chem. Soc., Dalton Trans. (8): 1475–1480. doi:10.1039/dt9960001475. ISSN 0300-9246.
- ↑ Boehme, Christian; Frenking, Gernot (1996-01-01). "Electronic Structure of Stable Carbenes, Silylenes, and Germylenes". Journal of the American Chemical Society. 118 (8): 2039–2046. doi:10.1021/ja9527075. ISSN 0002-7863.
- ↑ Schmedake, Thomas A.; Haaf, Michael; Apeloig, Yitzhak; Müller, Thomas; Bukalov, Sergey; West, Robert (1999-10-01). "Reversible Transformation between a Diaminosilylene and a Novel Disilene". Journal of the American Chemical Society. 121 (40): 9479–9480. doi:10.1021/ja992150j. ISSN 0002-7863.
- ↑ Li, Wenjian; Hill, Nicholas J.; Tomasik, Adam C.; Bikzhanova, Galina; West, Robert (2006-07-01). "A New Monomeric Saturated N-Heterocyclic Silylene as a Racemic Mixture". Organometallics. 25 (16): 3802–3805. doi:10.1021/om060544v. ISSN 0276-7333.
- ↑ Haaf, Michael; Schmiedl, Andreas; Schmedake, Thomas A.; Powell, Douglas R.; Millevolte, Anthony J.; Denk, Michael; West, Robert (1998-12-01). "Synthesis and Reactivity of a Stable Silylene". Journal of the American Chemical Society. 120 (49): 12714–12719. doi:10.1021/ja9733999. ISSN 0002-7863.
- ↑ Joo, Hyun; McKee, Michael L. (2005-04-01). "Computational Study of the "Stable" Bis(amino)silylene Reaction with Halomethanes. A Radical or Concerted Mechanism?". The Journal of Physical Chemistry A. 109 (16): 3728–3738. doi:10.1021/jp044458p. ISSN 1089-5639. PMID 16839041.
- ↑ Gehrhus, Barbara; Hitchcock, Peter B.; Lappert, Michael F. (1998-03-01). "The Thermally Stable Silylene Si[{N(CH2But)}2C6H4-1,2]: Reactivity toward CN Double Bonds". Organometallics. 17 (7): 1378–1382. doi:10.1021/om9709603. ISSN 0276-7333.
- ↑ Gehrhus, Barbara; Lappert, Michael F. (1998-03-01). "Reactions of the stable bis(amino)silylene with multiply bonded compounds". Polyhedron. 17 (5–6): 999–1000. doi:10.1016/s0277-5387(97)00206-4. ISSN 0277-5387.
- ↑ Metzler, Nils; Denk, Michael (1996-01-01). "Synthesis of a silylene–borane adduct and its slow conversion to a silylborane". Chem. Commun. (23): 2657–2658. doi:10.1039/cc9960002657. ISSN 1359-7345.
- ↑ Marco Boesveld, W.; Gehrhus, Barbara; Hitchcock, Peter B.; Lappert, Michael F.; von Ragué Schleyer, Paul (1999-01-01). "A crystalline carbene–silylene adduct 1,2-C6H4[N(R)]2C-Si[N(R)]2C6H4-1,2 (R = CH2But); synthesis, structure and bonding in model compounds†". Chemical Communications (8): 755–756. doi:10.1039/a900784i. ISSN 1359-7345.
- 1 2 3 Blom, Burgert; Stoelzel, Miriam; Driess, Matthias (2012-12-11). "New Vistas in N-Heterocyclic Silylene (NHSi) Transition-Metal Coordination Chemistry: Syntheses, Structures and Reactivity towards Activation of Small Molecules". Chemistry - A European Journal. 19 (1): 40–62. doi:10.1002/chem.201203072. ISSN 0947-6539. PMID 23229274.
- ↑ Petri, Stefan H. A.; Eikenberg, Dirk; Neumann, Beate; Stammler, Hans-Georg; Jutzi, Peter (1999-07-01). "Reaction of Molybdenocene and Tungstenocene Derivatives with the Divalent Silicon Species SiNtBuCHCHNtBu and (C5Me5)2Si". Organometallics. 18 (14): 2615–2618. doi:10.1021/om990115f. ISSN 0276-7333.
- ↑ Dysard, Jeffrey M.; Tilley, T. Don (2000-11-01). "Synthesis and Reactivity of (Pentamethylcyclopentadienyl)ruthenium Complexes of the Stable Neutral Silylene Si(tBuNCHCHNtBu)". Organometallics. 19 (23): 4726–4732. doi:10.1021/om000517j. ISSN 0276-7333.
- ↑ Zeller, Alexander; Bielert, Frank; Haerter, Peter; Herrmann, Wolfgang A.; Strassner, Thomas (2005-07-01). "Replacement of N-heterocyclic carbenes by N-heterocyclic silylenes at a Pd(0) center: Experiment and theory". Journal of Organometallic Chemistry. 690 (13): 3292–3299. doi:10.1016/j.jorganchem.2005.03.061. ISSN 0022-328X.
- ↑ Fürstner, Alois; Krause, Helga; Lehmann, Christian W. (2001-01-01). "Preparation, structure and catalytic properties of a binuclear Pd(0) complex with bridging silylene ligands". Chemical Communications (22): 2372. doi:10.1039/b108132b. ISSN 1359-7345. PMID 12240081.
- ↑ Zhang, Ming; Liu, Xiaoli; Shi, Chaojun; Ren, Chunxia; Ding, Yuqiang; Roesky, Herbert W. (2008-08-01). "The Synthesis of (η3-C3H5)Pd{Si[N(tBu)CH]2}Cl and the Catalytic Property for Heck Reaction". Zeitschrift für Anorganische und Allgemeine Chemie. 634 (10): 1755–1758. doi:10.1002/zaac.200800131. ISSN 0044-2313.