Quantum computational chemistry is an emerging field that integrates quantum mechanics with computational methods to simulate chemical systems. Despite quantum mechanics' foundational role in understanding chemical behaviors, traditional computational approaches face significant challenges, largely due to the complexity and computational intensity of quantum mechanical equations. This complexity arises from the exponential growth of a quantum system's wave function with each added particle, making exact simulations on classical computers inefficient.[1]
Efficient quantum algorithms for chemistry problems are expected to have run-times and resource requirements that scale polynomially with system size and desired accuracy. Experimental efforts have validated proof-of-principle chemistry calculations, though currently limited to small systems.[1]
Historical context for classical computational challenges in quantum mechanics
- 1929: Dirac noted the inherent complexity of quantum mechanical equations, underscoring the difficulties in solving these equations using classical computation.[2]
- 1982: Feynman proposed using quantum hardware for simulations, addressing the inefficiency of classical computers in simulating quantum systems.[3]
Methods in quantum complexity
Qubitization
One of the problems with hamiltonian simulation is the computational complexity inherent to its formation. Qubitization is a mathematical and algorithmic concept in quantum computing to the simulation of quantum systems via Hamiltonian dynamics. The core idea of qubitization is to encode the problem of Hamiltonian simulation in a way that is more efficiently processable by quantum algorithms.[4]
Qubitization involves a transformation of the Hamiltonian operator, a central object in quantum mechanics representing the total energy of a system. In classical computational terms, a Hamiltonian can be thought of as a matrix describing the energy interactions within a quantum system. The goal of qubitization is to embed this Hamiltonian into a larger, unitary operator, which is a type of operator in quantum mechanics that preserves the norm of vectors upon which it acts. This embedding is crucial for enabling the Hamiltonian's dynamics to be simulated on a quantum computer.[4]
Mathematically, the process of qubitization constructs a unitary operator such that a specific projection of proportional to the Hamiltonian H of interest. This relationship can often be represented as , where is a specific quantum state and is its conjugate transpose. The efficiency of this method comes from the fact that the unitary operator can be implemented on a quantum computer with fewer resources (like qubits and quantum gates) than would be required for directly simulating [4]





A key feature of qubitization is in simulating Hamiltonian dynamics with high precision while reducing the quantum resource overhead. This efficiency is especially beneficial in quantum algorithms where the simulation of complex quantum systems is necessary, such as in quantum chemistry and materials science simulations. Qubitization also develops quantum algorithms for solving certain types of problems more efficiently than classical algorithms. For instance, it has implications for the Quantum Phase Estimation algorithm, which is fundamental in various quantum computing applications, including factoring and solving linear systems of equations.[4]
Applications of qubization in chemistry
Gaussian orbital basis sets
In Gaussian orbital basis sets, phase estimation algorithms have been optimized empirically from to where is the number of basis sets. Advanced Hamiltonian simulation algorithms have further reduced the scaling, with the introduction of techniques like Taylor series methods and qubitization, providing more efficient algorithms with reduced computational requirements.[5]



Plane wave basis sets
Plane wave basis sets, suitable for periodic systems, have also seen advancements in algorithm efficiency, with improvements in product formula-based approaches and Taylor series methods.[4]
Quantum phase estimation in chemistry
Overview
Phase estimation, as proposed by Kitaev in 1996, identifies the lowest energy eigenstate ( ) and excited states ( ) of a physical Hamiltonian, as detailed by Abrams and Lloyd in 1999.[6] In quantum computational chemistry, this technique is employed to encode fermionic Hamiltonians into a qubit framework.[7]


Brief methodology
Initialization
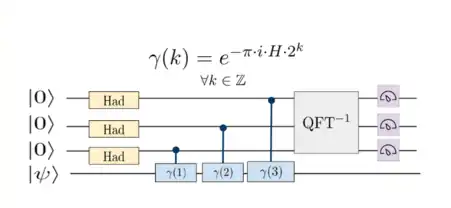




The qubit register is initialized in a state, which has a nonzero overlap with the Full Configuration Interaction (FCI) target eigenstate of the system. This state is expressed as a sum over the energy eigenstates of the Hamiltonian, , where represents complex coefficients.[9]


Application of Hadamard gates
Each ancilla qubit undergoes a Hadamard gate application, placing the ancilla register in a superposed state. Subsequently, controlled gates, as shown above, modify this state.[9]
Inverse quantum fourier transform
This transform is applied to the ancilla qubits, revealing the phase information that encodes the energy eigenvalues.[9]
Measurement
The ancilla qubits are measured in the Z basis, collapsing the main register into the corresponding energy eigenstate based on the probability .[9]

Requirements
The algorithm requires ancilla qubits, with their number determined by the desired precision and success probability of the energy estimate. Obtaining a binary energy estimate precise to n bits with a success probability necessitates.[9] ancilla qubits. This phase estimation has been validated experimentally across various quantum architectures.[9]



Applications of QPEs in chemistry
Time evolution and error analysis
The total coherent time evolution required for the algorithm is approximately .[10] The total evolution time is related to the binary precision , with an expected repeat of the procedure for accurate ground state estimation. Errors in the algorithm include errors in energy eigenvalue estimation (), unitary evolutions (), and circuit synthesis errors (), which can be quantified using techniques like the Solovay-Kitaev theorem.[11]






The phase estimation algorithm can be enhanced or altered in several ways, such as using a single ancilla qubit for sequential measurements, increasing efficiency, parallelization, or enhancing noise resilience in analytical chemistry. The algorithm can also be scaled using classically obtained knowledge about energy gaps between states.[12]
Limitations
Effective state preparation is needed, as a randomly chosen state would exponentially decrease the probability of collapsing to the desired ground state. Various methods for state preparation have been proposed, including classical approaches and quantum techniques like adiabatic state preparation.[13]
Variational quantum eigensolver (VQE)
Overview
The variational quantum eigensolver is an innovative algorithm in quantum computing, crucial for near-term quantum hardware.[14] Initially proposed by Peruzzo et al. in 2014 and further developed by McClean et al. in 2016, VQE is integral in finding the lowest eigenvalue of Hamiltonians, particularly those in chemical systems.[15] It employs the variational method (quantum mechanics), which guarantees that the expectation value of the Hamiltonian for any parameterized trial wave function is at least the lowest energy eigenvalue of that Hamiltonian.[16] This principle is fundamental in VQE's strategy to optimize parameters and find the ground state energy. VQE is a hybrid algorithm that utilizes both quantum and classical computers. The quantum computer prepares and measures the quantum state, while the classical computer processes these measurements and updates the system. This synergy allows VQE to overcome some limitations of purely quantum methods.[17]
Applications of VQEs in chemistry
1-RDM and 2-RDM calculations
The reduced density matrices (1-RDM and 2-RDM) can be used to extrapolate the electronic structure of a system.[18]
Ground state energy extrapolation
In the Hamiltonian variational ansatz, the initial state is prepared to represent the ground state of the molecular Hamiltonian without electron correlations. The evolution of this state under the Hamiltonian, split into commuting segments , is given by the equation bellow.[17]



where are variational parameters optimized to minimize the energy, providing insights into the electronic structure of the molecule.[17]

Measurement scaling
McClean et al. (2016) and Romero et al. (2019) proposed a formula to estimate the number of measurements ( ) required for energy precision. The formula is given by , where are coefficients of each Pauli string in the Hamiltonian. This leads to a scaling of in a Gaussian orbital basis and in a plane wave dual basis. Note that is the number of basis functions in the chosen basis set.[19][20]





Fermionic level grouping
A method by Bonet-Monroig, Babbush, and O'Brien (2019) focuses on grouping terms at a fermionic level rather than a qubit level, leading to a measurement requirement of only circuits with an additional gate depth of .[21]


Limitations of VQE
While VQE's application in solving the electronic Schrödinger equation for small molecules has shown success, its scalability is hindered by two main challenges: the complexity of the quantum circuits required and the intricacies involved in the classical optimization process.[22] These challenges are significantly influenced by the choice of the variational ansatz, which is used to construct the trial wave function. Consequently, the development of an efficient ansatz is a key focus in current research. Modern quantum computers face limitations in running deep quantum circuits, especially when using the existing ansatzes for problems that exceed several qubits.[17]
Jordan-Wigner encoding
Jordan-Wigner encoding is a fundamental method in quantum computing, extensively used for simulating fermionic systems like molecular orbitals and electron interactions in quantum chemistry.[23]
Overview
In quantum chemistry, electrons are modeled as fermions with antisymmetric wave functions. The Jordan-Wigner encoding maps these fermionic orbitals to qubits, preserving their antisymmetric nature. Mathematically, this is achieved by associating each fermionic creation and annihilation operator with corresponding qubit operators through the Jordan-Wigner transformation:



Where , , and are Pauli matrices acting on the qubit.[23]




Applications of Jordan-Wigner encoding in chemistry
Electron hopping
Electron hopping between orbitals, central to chemical bonding and reactions, is represented by terms like . Under Jordan-Wigner encoding, these transform as follows:[23]


This transformation captures the quantum mechanical behavior of electron movement and interaction within molecules.[24]
Computational complexity in molecular systems
The complexity of simulating a molecular system using Jordan-Wigner encoding is influenced by the structure of the molecule and the nature of electron interactions. For a molecular system with orbitals, the number of required qubits scales linearly with , but the complexity of gate operations depends on the specific interactions being modeled.[25]

Limitations of Jordan–Wigner encoding
The Jordan-Wigner transformation encodes fermionic operators into qubit operators, but it introduces non-local string operators that can make simulations inefficient. The FSWAP gate is used to mitigate this inefficiency by rearranging the ordering of fermions (or their qubit representations), thus simplifying the implementation of fermionic operations.[26]
Fermionic SWAP (FSWAP) network
FSWAP networks rearrange qubits to efficiently simulate electron dynamics in molecules. These networks are essential for reducing the gate complexity in simulations, especially for non-neighboring electron interactions.[27]
When two fermionic modes (represented as qubits after the Jordan-Wigner transformation) are swapped, the FSWAP gate not only exchanges their states but also correctly updates the phase of the wavefunction to maintain fermionic antisymmetry. This is in contrast to the standard SWAP gate, which does not account for the phase change required in the antisymmetric wavefunctions of fermions.[28]
The use of FSWAP gates can significantly reduce the complexity of quantum circuits for simulating fermionic systems. By intelligently rearranging the fermions, the number of gates required to simulate certain fermionic operations can be reduced, leading to more efficient simulations. This is particularly useful in simulations where fermions need to be moved across large distances within the system, as it can avoid the need for long chains of operations that would otherwise be required.[29]
Interpreting solutions to molecular wavefunctions
The atoms in molecules (QTAIM) model of Richard Bader was developed to effectively link the quantum mechanical model of a molecule, as an electronic wavefunction, to chemically useful concepts such as atoms in molecules, functional groups, bonding, the theory of Lewis pairs, and the valence bond model. Bader has demonstrated that these empirically useful chemistry concepts can be related to the topology of the observable charge density distribution, whether measured or calculated from a quantum mechanical wavefunction. QTAIM analysis of molecular wavefunctions is implemented, for example, in the AIMAll software package.[30]
References
- 1 2 Gieres, François (2000). "Mathematical surprises and Dirac's formalism in quantum mechanics". Reports on Progress in Physics. 63 (12): 1893–1931. arXiv:quant-ph/9907069. Bibcode:2000RPPh...63.1893G. doi:10.1088/0034-4885/63/12/201. S2CID 250880658.
- ↑ Dirac, P. A. M. (1929-04-06). "Quantum mechanics of many-electron systems". Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character. 123 (792): 714–733. Bibcode:1929RSPSA.123..714D. doi:10.1098/rspa.1929.0094. ISSN 0950-1207. S2CID 121992478.
- ↑ Feynman, Richard P. (2019-06-17). Hey, Tony; Allen, Robin W. (eds.). Feynman Lectures On Computation. Boca Raton: CRC Press. doi:10.1201/9780429500442. ISBN 978-0-429-50044-2. S2CID 53898623.
- 1 2 3 4 5 Low, Guang Hao; Chuang, Isaac L. (2019-07-12). "Hamiltonian Simulation by Qubitization". Quantum. 3: 163. arXiv:1610.06546. Bibcode:2019Quant...3..163L. doi:10.22331/q-2019-07-12-163. S2CID 119109921.
- ↑ Kwon, Hyuk‐Yong; Curtin, Gregory M.; Morrow, Zachary; Kelley, C. T.; Jakubikova, Elena (2023-07-15). "Adaptive basis sets for practical quantum computing". International Journal of Quantum Chemistry. 123 (14). arXiv:2211.06471. doi:10.1002/qua.27123. ISSN 0020-7608. S2CID 253510818.
- ↑ Kitaev, Alexei (1996-01-17). Quantum measurements and the Abelian Stabilizer Problem (Report). Electronic Colloquium on Computational Complexity (ECCC).
- ↑ Abrams, Daniel S.; Lloyd, Seth (1999-12-13). "Quantum Algorithm Providing Exponential Speed Increase for Finding Eigenvalues and Eigenvectors". Physical Review Letters. 83 (24): 5162–5165. arXiv:quant-ph/9807070. Bibcode:1999PhRvL..83.5162A. doi:10.1103/PhysRevLett.83.5162. S2CID 118937256.
- ↑ McArdle, Sam; Endo, Suguru; Aspuru-Guzik, Alán; Benjamin, Simon C.; Yuan, Xiao (2020-03-30). "Quantum computational chemistry". Reviews of Modern Physics. 92 (1): 015003. arXiv:1808.10402. Bibcode:2020RvMP...92a5003M. doi:10.1103/RevModPhys.92.015003. S2CID 119476644.
- 1 2 3 4 5 6 Nielsen, Michael A.; Chuang, Isaac L. (2010). Quantum computation and quantum information (10th anniversary ed.). Cambridge: Cambridge university press. ISBN 978-1-107-00217-3.
- ↑ Du, Jiangfeng; Xu, Nanyang; Peng, Xinhua; Wang, Pengfei; Wu, Sanfeng; Lu, Dawei (2010-01-22). "NMR Implementation of a Molecular Hydrogen Quantum Simulation with Adiabatic State Preparation". Physical Review Letters. 104 (3): 030502. Bibcode:2010PhRvL.104c0502D. doi:10.1103/PhysRevLett.104.030502. ISSN 0031-9007. PMID 20366636.
- ↑ Lanyon, B. P.; Whitfield, J. D.; Gillett, G. G.; Goggin, M. E.; Almeida, M. P.; Kassal, I.; Biamonte, J. D.; Mohseni, M.; Powell, B. J.; Barbieri, M.; Aspuru-Guzik, A.; White, A. G. (2010). "Towards quantum chemistry on a quantum computer". Nature Chemistry. 2 (2): 106–111. arXiv:0905.0887. Bibcode:2010NatCh...2..106L. doi:10.1038/nchem.483. ISSN 1755-4349. PMID 21124400. S2CID 640752.
- ↑ Wang, Youle; Zhang, Lei; Yu, Zhan; Wang, Xin (2023). "Quantum Phase Processing and its Applications in Estimating Phase and Entropies". Physical Review A. 108 (6): 062413. arXiv:2209.14278. Bibcode:2023PhRvA.108f2413W. doi:10.1103/PhysRevA.108.062413.
- ↑ Sugisaki, Kenji; Toyota, Kazuo; Sato, Kazunobu; Shiomi, Daisuke; Takui, Takeji (2022-07-25). "Adiabatic state preparation of correlated wave functions with nonlinear scheduling functions and broken-symmetry wave functions". Communications Chemistry. 5 (1): 84. doi:10.1038/s42004-022-00701-8. ISSN 2399-3669. PMC 9814591. PMID 36698020.
- ↑ Peruzzo, Alberto; McClean, Jarrod; Shadbolt, Peter; Yung, Man-Hong; Zhou, Xiao-Qi; Love, Peter J.; Aspuru-Guzik, Alán; O'Brien, Jeremy L. (2014-07-23). "A variational eigenvalue solver on a photonic quantum processor". Nature Communications. 5 (1): 4213. arXiv:1304.3061. Bibcode:2014NatCo...5.4213P. doi:10.1038/ncomms5213. ISSN 2041-1723. PMC 4124861. PMID 25055053.
- ↑ Peruzzo, Alberto; McClean, Jarrod; Shadbolt, Peter; Yung, Man-Hong; Zhou, Xiao-Qi; Love, Peter J.; Aspuru-Guzik, Alán; O’Brien, Jeremy L. (2014-07-23). "A variational eigenvalue solver on a photonic quantum processor". Nature Communications. 5 (1): 4213. arXiv:1304.3061. Bibcode:2014NatCo...5.4213P. doi:10.1038/ncomms5213. ISSN 2041-1723. PMC 4124861. PMID 25055053.
- ↑ Chan, Albie; Shi, Zheng; Dellantonio, Luca; Dur, Wolfgang; Muschik, Christine A (2023). "Hybrid variational quantum eigensolvers: merging computational models". arXiv:2305.19200 [quant-ph].
- 1 2 3 4 Tilly, Jules; Chen, Hongxiang; Cao, Shuxiang; Picozzi, Dario; Setia, Kanav; Li, Ying; Grant, Edward; Wossnig, Leonard; Rungger, Ivan; Booth, George H.; Tennyson, Jonathan (2022-11-05). "The Variational Quantum Eigensolver: A review of methods and best practices". Physics Reports. 986: 1–128. arXiv:2111.05176. Bibcode:2022PhR...986....1T. doi:10.1016/j.physrep.2022.08.003. ISSN 0370-1573. S2CID 243861087.
- ↑ Liu, Jie; Li, Zhenyu; Yang, Jinlong (2021-06-28). "An efficient adaptive variational quantum solver of the Schrödinger equation based on reduced density matrices". The Journal of Chemical Physics. 154 (24). arXiv:2012.07047. Bibcode:2021JChPh.154x4112L. doi:10.1063/5.0054822. ISSN 0021-9606. PMID 34241330. S2CID 229156865.
- ↑ Romero, Jonathan; Babbush, Ryan; McClean, Jarrod R; Hempel, Cornelius; Love, Peter J; Aspuru-Guzik, Alán (2018-10-19). "Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz". Quantum Science and Technology. 4 (1): 014008. arXiv:1701.02691. doi:10.1088/2058-9565/aad3e4. ISSN 2058-9565. S2CID 4175437.
- ↑ McClean, Jarrod R; Romero, Jonathan; Babbush, Ryan; Aspuru-Guzik, Alán (2016-02-04). "The theory of variational hybrid quantum-classical algorithms". New Journal of Physics. 18 (2): 023023. arXiv:1509.04279. Bibcode:2016NJPh...18b3023M. doi:10.1088/1367-2630/18/2/023023. ISSN 1367-2630. S2CID 92988541.
- ↑ Bonet-Monroig, Xavier; Babbush, Ryan; O'Brien, Thomas E. (2020-09-22). "Nearly Optimal Measurement Scheduling for Partial Tomography of Quantum States". Physical Review X. 10 (3): 031064. arXiv:1908.05628. Bibcode:2020PhRvX..10c1064B. doi:10.1103/PhysRevX.10.031064. S2CID 199668962.
- ↑ Grimsley, Harper R.; Barron, George S.; Barnes, Edwin; Economou, Sophia E.; Mayhall, Nicholas J. (2023-03-01). "Adaptive, problem-tailored variational quantum eigensolver mitigates rough parameter landscapes and barren plateaus". npj Quantum Information. 9 (1): 19. arXiv:2204.07179. Bibcode:2023npjQI...9...19G. doi:10.1038/s41534-023-00681-0. ISSN 2056-6387. S2CID 257236255.
- 1 2 3 Jiang, Zhang; Sung, Kevin J.; Kechedzhi, Kostyantyn; Smelyanskiy, Vadim N.; Boixo, Sergio (2018-04-26). "Quantum Algorithms to Simulate Many-Body Physics of Correlated Fermions". Physical Review Applied. 9 (4): 044036. arXiv:1711.05395. Bibcode:2018PhRvP...9d4036J. doi:10.1103/PhysRevApplied.9.044036. ISSN 2331-7019. S2CID 54064506.
- ↑ Li, Qing-Song; Liu, Huan-Yu; Wang, Qingchun; Wu, Yu-Chun; Guo, Guo-Ping (2022). "A unified framework of transformations based on the Jordan–Wigner transformation". The Journal of Chemical Physics. 157 (13). arXiv:2108.01725. Bibcode:2022JChPh.157m4104L. doi:10.1063/5.0107546. PMID 36209000. S2CID 236912625. Retrieved 2023-11-13.
- ↑ Harrison, Brent; Nelson, Dylan; Adamiak, Daniel; Whitfield, James (November 2022). "Reducing the qubit requirement of Jordan-Wigner encodings of N-mode, K-fermion systems from N to ⌈log2(NK)⌉". arXiv:2211.04501 [quant-ph].
- ↑ "Custom Fermionic Codes for Quantum Simulation | Perimeter Institute". www2.perimeterinstitute.ca. Retrieved 2023-11-13.
- ↑ Kivlichan, Ian D.; McClean, Jarrod; Wiebe, Nathan; Gidney, Craig; Aspuru-Guzik, Alán; Chan, Garnet Kin-Lic; Babbush, Ryan (2018-03-13). "Quantum Simulation of Electronic Structure with Linear Depth and Connectivity". Physical Review Letters. 120 (11): 110501. arXiv:1711.04789. Bibcode:2018PhRvL.120k0501K. doi:10.1103/PhysRevLett.120.110501. PMID 29601758. S2CID 4219888.
- ↑ Hashim, Akel; Rines, Rich; Omole, Victory; Naik, Ravi K; John MarkKreikebaum, John Mark; Santiago, David I; Chong, Frederic T.; Siddiqi, Irfan; Gokhale, Pranav (2021). "Optimized fermionic SWAP networks with equivalent circuit averaging for QAOA". arXiv:2111.04572 [quant-ph].
- ↑ Rubin, Nicholas C.; Gunst, Klaas; White, Alec; Freitag, Leon; Throssell, Kyle; Chan, Garnet Kin-Lic; Babbush, Ryan; Shiozaki, Toru (2021-10-27). "The Fermionic Quantum Emulator". Quantum. 5: 568. arXiv:2104.13944. Bibcode:2021Quant...5..568R. doi:10.22331/q-2021-10-27-568. S2CID 233443911.
- ↑ Bader, Richard F. W. (2003). Atoms in molecules: a quantum theory. The international series of monographs on chemistry (Reprinted ed.). Oxford: Clarendon Press. ISBN 978-0-19-855865-1.