Spatial transcriptomics is a method for assigning cell types (identified by the mRNA readouts) to their locations in the histological sections and can also be used to determine subcellular localization of mRNA molecules. First described in 2016 by Ståhl et al., it has since undergone a variety of improvements and modifications.[1]
The Ståhl method[1] implies positioning individual tissue samples on the arrays of spatially barcoded reverse transcription primers able to capture mRNA with the oligo(dT) tails.[1] Besides oligo(dT) tail and spatial barcode, which indicates the x and y position on the arrayed slide, the probe contains a cleavage site, amplification and sequencing handle, and unique molecular identifier.[1] Commonly, histological samples are cut using cryotome, then fixed, stained, and put on the microarrays.[1] After that, it undergoes enzymatic permeabilization, so that molecules can diffuse down to the slide, with further mRNA release and binding to the probes.[1] Reverse transcription is then carried out in situ.[1] As a result, spatially marked complementary DNA is synthesized, providing information about gene expression in the exact location of the sample.[1] Thus, described protocol combines paralleled sequencing and staining of the same sample.[1] It is important to mention that the first generation of the arrayed slides comprised about 1,000 spots of the 100-μm diameter, limiting resolution to ~10-40 cells per spot.[1]
In the broader meaning of this term, spatial transcriptomics includes methods that can be divided into five principal approaches[2] to resolving spatial distribution of transcripts. They are microdissection techniques, Fluorescent in situ hybridization methods, in situ sequencing, in situ capture protocols and in silico approaches.[2]
Application
Defining the spatial distribution of mRNA molecules allows for the experimentalist to uncover cellular heterogeneity in tissues, tumours, immune cells as well as determine the subcellular distribution of transcripts in various conditions.[2] This information provides a unique opportunity to decipher both the cellular and subcellular architecture in both tissues and individual cells. These methodologies provide crucial insights in the fields of embryology, oncology, immunology and histology.[2] The functioning of the individual cells in multicellular organisms can only be completely explained in the context of identifying their exact location in the body.[2] Spatial transcriptomics techniques sought to elucidate cells’ properties this way. Below, we look into the methods that connect gene expression to the spatial organization of cells.[2]
Microdissection
Laser capture microdissection
Laser capture microdissection enables capturing single cells without causing morphologic alterations.[4][5] It exploits transparent ethylene vinyl acetate film apposed to the histological section and a low-power infrared laser beam.[4] Once such beam is directed at the cells of interest, film directly above the targeted area temporarily melts so that its long-chain polymers cover and tightly capture the cells.[4] Then, the section is removed and cells of interest remain embedded in the film.[4] This method allows further RNA transcript profiling and cDNA library generation of the retrieved cells.
RNA sequencing of individual cryosections
RNA sequencing of the selected regions in individual cryosections is another method that can produce location-based genome-wide expression data.[6] This method is carried out without laser capture microdissection. It was first used to determine genome-wide spatial patterns of gene expression in cryo-sliced Drosophila embryos.[6] Essentially, it implies simple preparation of the library from the selected regions of the sample. This method had difficulties in obtaining high-quality RNA-seq libraries from every section due to the material loss as a result of the small amount of total RNA in each slice.[2][6] This problem was resolved by adding RNA of a distantly related Drosophila species to each tube after initial RNA extraction.[6]
TIVA
Transcriptome in vivo analysis (TIVA) is a technique that enables capturing mRNA in live single cells in intact live tissue sections.[7] It uses a photoactivatable tag.[2][7] The TIVA tag has several functional groups and a trapped poly(U) oligonucleotide coupled to biotin.[7] A disulfide-linked peptide, which is adjacent to the tag, allows it to penetrate the cell membrane.[7] Once inside, laser photoactivation is used to unblock poly(U) oligonucleotide in the cells of interest, so that TIVA tag hybridizes to mRNAs within the cell.[7] Then, streptavidin capture of the biotin group is used to extract poly(A)-tailed mRNA molecules bound to unblocked tags, after which these mRNAs are analyzed by RNA sequencing.[7] This method is limited by low throughput, as only a few single cells can be processed at a time.[2]
tomo-seq
An advanced alternative for RNA Sequencing of Individual Cryosections described above, RNA tomography (tomo-seq) features better RNA quantification and spatial resolution.[8] It is also based on tissue cryosectioning with further RNA sequencing of individual sections, yielding genome-wide expression data and preserving spatial information.[8] In this protocol, usage of carrier RNA is omitted due to linear amplification of cDNA in individual histological sections.[8] The identical sample is sectioned in different directions followed by 3D transcriptional construction using overlapping data.[8] Overall, this method implies using identical samples for each section and thus cannot be applied for processing clinical material.[2]
LCM-seq
LCM-seq utilizes laser capture microdissection (LCM) coupled with Smart-Seq2 RNA sequencing and is applicable down to the single cell level and can even be used on partially degraded tissues. The workflow includes cryosectioning of tissues followed by laser capture microdissection, where cells are collected directly into lysis buffer and cDNA is generated without the need for RNA isolation, which both simplifies the experimental procedures as well as lowers technical noise.[9][10] As the positional identity of each cell is recorded during the LCM procedure, the transcriptome of each cell after RNA sequencing of the corresponding cDNA library can be inferred to the position where it was isolated from.[9] LCM-seq has been applied to multiple cell types to understand their intrinsic properties, including oculomotor neurons, facial motor neurons, hypoglossal motor neurons, spinal motor neurons, red nucleus neurons,[11] interneurons,[12] dopamine neurons,[13] and chondrocytes.[14]
Geo-seq
Geo-seq is a method that utilizes both laser capture microdissection and single-cell RNA sequencing procedures to determine the spatial distribution of the transcriptome in tissue areas approximately ten cells in size.[15] The workflow involves removal and cryosectioning of tissue followed by laser capture microdissection.[15][16] The extracted tissue is then lysed, and the RNA is purified and reverse transcribed into a cDNA library.[15][16] Library is sequenced, and the transcriptomic profile can be mapped to the original location of the extracted tissue.[15][16] This technique allows the user to define regions of interest in a tissue, extract said tissue and map the transcriptome in a targeted approach.
NICHE-seq
The NICHE-seq method uses photoactivatable fluorescent markers and two-photon laser scanning microscopy to provide spatial data to the transcriptome generated.[17] The cells bound by the fluorescent marker are photoactivated, dissociated and sorted via fluorescence-activated cell sorting.[17] This provides sorting specificity to only labeled, photoactivated cells.[17] Following sorting, single-cell RNA sequencing generates the transcriptome of the visualized cells.[17] This method can process thousands of cells within a defined niche at the cost of losing spatial data between cells in the niche.[17]
ProximID
ProximID is a methodology based on iterative micro digestion of extracted tissue to single cells.[18] Initial mild digestion steps preserve small interacting structures that are recorded prior to continued digestion.[18] The single cells are then separated from each structure and undergo sc-RNAseq and clustered using t-distributed stochastic neighbour embedding.[18][19] The clustered cells can be mapped to physical interactions based on the interacting structures prior to the micro digestions.[18] While the throughput of this technique is relatively low it provides information on physical interaction between cells to the dataset.[2]
Fluorescent in situ hybridization
NanoString CosMx
CosMx Spatial Molecular Imager is the first high-plex in situ analysis platform to provide spatial multiomics with formalin-fixed paraffin-embedded (FFPE) and fresh frozen (FF) tissue samples at cellular and subcellular resolution. It enables rapid quantification and visualization of up to 1,000 RNA and 64 validated protein analytes and is the flexible, spatial single-cell imaging platform for cell atlasing, tissue phenotyping, cell-cell interactions, cellular processes, and biomarker discovery.
smFISH
One of the first techniques able to achieve spatially resolved RNA profiling of individual cells was single-molecule fluorescent in situ hybridization (smFISH).[20] It implemented short (50 base pairs) oligonucleotide probes conjugated with 5 fluorophores which could bind to a specific transcript yielding bright spots in the sample.[20] Detection of these spots provides quantitative information about expression of certain genes in the cell.[20] However, usage of probes labeled with multiple fluorophores was challenged by self-quenching, altered hybridization characteristics, their synthesis and purification.[2]
Later, this method was changed[21] by substituting the above described probes with those of 20 bp length, coupled to only one fluorophore and complementary in tandem to an mRNA sequence of interest, meaning that those would collectively bind to the targeted mRNA. One such probe itself wouldn't produce a strong signal, but the cumulative fluorescence of the congregated probes would show a bright spot. Since single misbound probes are unlikely to co-localize, false-positive signals in this method are limited.[2]
Thus, this in situ hybridization (ISH) technique spots spatial localization of RNA expression via direct imaging of individual RNA molecules in single cells.
RNAscope
Another in situ hybridization technique termed RNAscope employs probes of the specific Z-shaped design to simultaneously amplify hybridization signals and suppress background noise.[22] It allows for the visualization of single RNA in a variety of cellular types.[23] Most steps of RNAScope are similar to the classic ISH protocol.[22] The tissue sample is fixed onto slides and then treated with RNAscope reagents that permeate the cells. Z-probes are designed in a way that they are only effective when bound in pairs to the target sequence.[22] This allows another element of this method (preamplifier) to connect to the top tails of Z-probes.[22] Once affixed, preamplifier serves as a binding site for other elements: amplifiers which in turn bind to another type of probes: label probes.[22] As a result, a bulky structure is formed on the target sequence. Most importantly, the preamplifier fails to bind to a singular Z-probe, thus, nonspecific binding wouldn't entail signal emission, thus, eliminating background noise mentioned in the beginning.[2][22]
seqFISH
Sequential fluorescence in situ hybridization (seqFISH) is another method that provides identification of mRNA directly in single cells with preservation of their spatial context.[24][25] This method is carried out in multiple rounds; each of them includes fluorescent probe hybridization, imaging, and consecutive probe stripping.[24] Various genes are assigned different colors in every round, generating a unique temporal barcode.[24] Thus, seqFISH distinguishes mRNAs by a sequential color code, such as red-red-green. Nevertheless, this technique has its flaws featuring autofluorescent background and high costs due to the number of probes used in each round.[2]
MERFISH
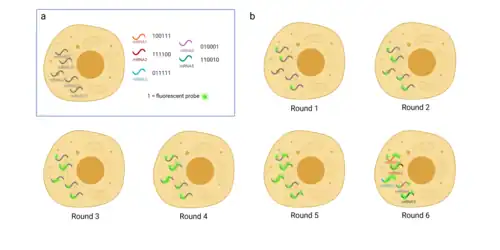
Conventional FISH methods are limited by the small number of genes that can be simultaneously analyzed due to the small number of distinct color channels, so multiplexed error-robust FISH was designed to overcome this problem.[26] Multiplexed Error-Robust FISH (MERFISH) greatly increases the number of RNA species that can be simultaneously imaged in single cells employing binary code gene labeling in multiple rounds of hybridization.[27] This approach can measure 140 RNA species at a time using an encoding scheme that both detects and corrects errors.[27] The core principle lies in identification of genes by combining signals from several consecutive hybridization rounds and assigning N-bit binary barcodes to genes of interest.[27] The Code depends on specific probes and comprises “1” or “0” values and their combination is set differently for each gene.[27] Errors are avoided by using six-bit or longer codes with any two of them differing by at least 3 bits.[27] A specific probe is created for each RNA species.[27] Each probe is a target-specific oligonucleotide that consists of 20-30 base pairs and complementary binds to mRNA sequence after permeating the cell.[27] Then, multiple rounds of hybridization are conducted as follows: for each round, only a probe that includes “1” in the corresponding binary code position is added.[27] At the end of each round, fluorescent microscopy is used to locate each probe.[27] Expectedly, only those mRNAs which had “1” in the assigned position would be captured.[27] Photos are then photobleached and a new subset is added.[27] Thus, we retrieve combination of binary values which makes it possible to distinguish between numerous RNA species.
smHCR
Single-molecule RNA detection at depth by hybridization chain reaction (smHCR) is an advanced seqFISH technique that can overcome typical complication of autofluorescent background in thick and opaque tissue samples.[28] In this method, multiple readout probes are bound with the target region of mRNA.[28] Target is detected by a set of short DNA probes which attach to it in defined subsequence.[28] Each DNA probe carries an initiator for the same HCR amplifier.[28] Then, fluorophore-labeled DNA HCR hairpins penetrate the sample and assemble into fluorescent amplification polymers attaching to initiating probes.[28] In multiplexed studies, the same two-stage protocol described above is used: all probe sets are introduced simultaneously, just as all HCR amplifiers are; spectrally distinct fluorophores are used for further imaging.[28]
osmFISH
Cyclic-ouroboros smFISH (osmFISH) is an adaptation of smFISH which aims to overcome the challenge of optical crowding.[29] In osmFISH, transcripts are visualized, and an image is acquired before the probe is stripped and a new transcript is visualized with a different fluorescent probe.[29] After successive rounds the images are compiled to view the spatial distribution of the RNA.[29] Due to transcripts being sequentially visualized it eliminates the issue of signals interfering with each other.[2][29] This method allows the user to generate high resolution images of larger tissue sections than other related techniques.[2]
ExFISH
Expansion FISH (ExFISH) leverages expansion microscopy to allow for super-resolution imaging of RNA location, even in thick specimens such as brain tissue.[30] It supports both single-molecule and multiplexed readouts.[30]
EASI-FISH
Expansion-Assisted Iterative Fluorescence In Situ Hybridization (EASI-FISH) optimizes and builds on ExFISH with improved detection accuracy and robust multi-round processing across samples thicker (300 μm) than what was previously possible.[31] It also includes a turn-key computational analysis pipeline.[32]
seqFISH+
SeqFISH+ resolved optical issues related to spatial crowding by subsequent rounds of fluorescence.[33] First, a primary probe anneals to targeted mRNA and then subsequent probes bind to flanking regions of the primary probe resulting in a unique barcode.[33] Each readout probe is captured as an image and collapsed into a super resolved image.[33] This method allows the user to target up to ten thousand genes at a time.[2]
DNA microscopy
DNA microscopy is a distinct imaging method for optics-free mapping of molecules’ positions with simultaneous preservation of sequencing data carried out in several consecutive in situ reactions.[34] First, cells are fixed and cDNA is synthesized.[34] Randomized nucleotides then tag target cDNAs in situ, providing unique labels for each molecule.[34] Tagged transcripts are amplified in the second in situ reaction, retrieved copies are concatenated, and new randomized nucleotides are added.[34] Each consecutive concatenation event is labeled, yielding unique event identifiers.[34] Algorithm then generates images of the original transcripts based on decoded molecular proximities from the obtained concatenated sequences, while target's single nucleotide information is being recorded as well.[34]
in situ sequencing
ISS using padlock probes
The ISS padlock method[35] is based on padlock probing,[36] rolling-circle amplification (RCA),[37] and sequencing by ligation chemistry.[38] Within intact tissue sections, mRNA is reversely transcribed to cDNA, which is followed by mRNA degradation by RNase H.[2] Then, there are two ways of how this method can be carried out. The first way, gap-targeted sequencing, involves padlock probe binding to cDNA with a gap between the ends of the probe which are targeted for sequencing by ligation.[2] DNA polymerization then fills this gap and a DNA circle is created by DNA ligation.[2] Another way, barcode-targeted sequencing, DNA circularization of a padlock probe with a barcode sequence is conducted by ligation only.[2] In both versions of the method, the ends are ligated forming a circle of DNA.[2] Target amplification is then performed by RCA, yielding micrometer-sized RCA products (RCPs).[2] RCAs consist of repeats of the padlock probe sequence.[2] These DNA molecules are then subjected to sequencing by ligation, decoding either a gap-filled sequence or an up to four-base-long barcode within the probe with adjacent ends, depending on the version.[2] No-gap variant claims higher sensitivity, while gap-filled one implies reading out the actual RNA sequence of the transcript.[2] Later, this method was improved by automatization on a microfluidic platform and substitution of sequencing by ligation with sequencing by hybridization technology.[2]
FISSEQ
Fluorescent in situ sequencing (FISSEQ),[39] like ISS padlock, is a method that uses reverse transcription, rolling-circle amplification, and sequencing by ligation techniques.[2] It allows spatial transcriptome analysis in fixed cells.[2] RNA is first reverse transcribed into cDNA with regular and modified amine-bases and tagged random hexamer RT primers.[2] Amine-bases mediate the cross-linkage of cDNA to its cellular surrounding.[2] Then cDNA is circulated by ligation and amplified by RCA.[2] Single-stranded DNA nanoballs of 200–400 nm in diameter are obtained as a result.[2] Thus, these nanoballs comprise numerous tandem repeats of the cDNA sequence. Then sequencing is performed via SOLiD sequencing by ligation.[2] Positions of both product of reverse transcription and clonally amplified RCPs are maintained via cross-linkage to cellular matrix components mentioned previously, creating a 3D in situ RNA-seq library within the cell.[2] Once bound with fluorescent probes featuring different colors, amplicons become highly fluorescent which allows visual detection of the signal; however, the image-processing algorithm relies on read alignment to reference sequences rather than signal intensity.[2]
Barista-seq
Barcode in situ targeted sequencing (Barista-seq) is an improvement on the gap padlock probe methodology boasting a fivefold increase in efficiency, an increased read length of fifteen bases and is compatible with illumina sequencing platforms.[2][19] The method also uses padlock probes and rolling circle amplification, however this approach uses sequencing-by-synthesis and crosslinking unlike the gap padlock method.[19] The crosslinking to the cellular matrix in the same procedure is the same as FISSEQ.[2][19]
STARmap
Spatially-resolved transcript amplicon readout mapping (STARmap) utilizes a padlock probe with an additional primer which allows for direct amplification of mRNA, forgoing the need for reverse transcription.[2][40] Similar to other padlock probe based methods amplification occurs via rolling circle amplification.[2] The DNA amplicons are chemically modified and embedded into a polymerized hydrogel within the cell.[2] Captured RNA can then be sequenced in situ providing three dimensional locations of the mRNA within each cell.[2]
in situ capture
Stereo-seq(STOmics)
STOmics is a pioneer in advancing spatially-resolved transcriptomic analysis through its proprietary SpaTial Enhanced REsolution Omics-Sequencing (Stereo-seq) technology.[41]
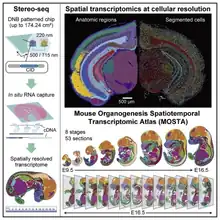
It combines in situ capture with DNB-seq, DNB sequencing is based on lithographically etched chips (patterned arrays) for in situ sequencing. Unlike other um-level in situ capture technologies, standard DNB chips have spots with approximately 220 nm diameter and a center-to-center distance of 500 nm, providing up to 20000 spots for tissue RNA capture per 10mm linear distance, or 4x108 spots per 1cm2. Therefore, STOmics can show higher resolution and wider field of view than other in situ capture technologies.[42]
Spatial transcriptomics
Spatial Transcriptomics is the ability to capture the positional context of transcriptional activity within intact tissue, either for regions or single cells.
When a tissue cryosection is attached to a spatial transcriptomic slide the barcoded primers bind and capture adjacent mRNAs from the tissue. While the tissue section is attached to the slide, reverse transcription of captured mRNA is initiated and the resulting cDNA incorporates the spatial barcode of the primer. Following mRNA capture and reverse transcription, sequencing libraries are prepared and analyzed with Illumina dye sequencing. The spatial barcode present within each generated sequence allows the data for each individual mRNA transcript to be mapped back to its point of origin within the tissue section.
NanoString GeoMx
NanoString's GeoMx Digital Spatial Profiler (DSP) allows the user to define a microscopic region of interest on an FFPE or frozen tissue slide due to a UV-photocleavable barcode engineered into the in-situ hybridization probes.[2][43] The region of interest is specifically exposed to UV light, the barcodes are cleaved and used to identify the RNA or protein present in the tissue.[43] The defined regions of interest can vary in size between ten and six hundred micrometers allowing targeting of a wide variety of structures and cells in the histological sample.[2]
Slide-seq
Slide-seq relies on the attachment of RNA binding, DNA-barcoded micro beads to a rubber coated glass coverslip.[2][44] The microbeads are mapped to their spatial location via SOLiD sequencing.[44] Tissue sections are transferred to this coverslip to capture extracted RNA. Captured RNA is amplified and sequenced.[44] Transcript localization is determined by the barcode oligonucleotide sequence from the bead that captured it.[44]
APEX-seq
APEX-seq allows the for assessment of the spatial transcriptome in different regions of a cell.[2][45] The method utilizes the APEX2 gene, expressed in live cells which are incubated with biotin-phenol and hydrogen peroxide.[45] In these conditions the APEX2 enzymes catalyse the transfer of biotin groups to the RNA molecules and these can then be purified via streptavidin bead purification.[45] The purified transcripts are then sequenced to determine which molecules were in close proximity to the biotin tagging enzyme.[45]
HDST
High-Definition Spatial Transcriptomics (HDST) begins with decoding the location of mRNA capture beads in wells on a glass slide.[46] This is accomplished by sequential hybridization to the barcode oligonucleotide sequence of each bead.[46] Once the location of each bead is decoded, a tissue sample can be placed on the slide and permeabilized.[46] The captured transcripts are then sequenced.[46] HDST uses smaller beads than Slide-seq and thus can resolve at a spatial resolution of two micrometers compared to ten micrometers of Slide-seq.[2]
10X Visium
The 10X Visium assay is a newer and improved version of the Spatial Transcriptomics assay. It also utilizes spotted arrays of mRNA-capturing probes on the surface of glass slides but with increased spot number, minimized spot size and increased amount of capture probes per spot. Within each of the four capture areas of the Visium Spatial Gene Expression slides, there are approximately 5000 barcoded spots, which in turn contain millions of spatially barcoded capture oligonucleotides. Tissue mRNA is released upon permeabilization and binds to the barcoded oligos, enabling capture of gene expression information. Each barcoded spot is 55 µm in diameter, and the distance from the center of one spot to the center of another is approximately 100 µm. The spots are staggered to minimize the distance between them. On average, mRNA from anywhere between 1 and 10 cells are captured per spot which provides near single-cell resolution. [47]
in silico construction
Reconstruction using ISH
in silico Spatial Reconstruction with ISH implies computational spatial reconstruction of cells’ locations according to their expression profiles.[2] Several similar methods of this principle exist.[48][49] They co-analyze single-cell transcriptomics and available ISH-based gene expression atlases of the same cell type.[2] Based on these data, cells are then assigned to their positions in the tissue. Obviously, this method is limited by the factor of availability of ISH references.[2] Additionally, it becomes more complicated when assigning cells in complex tissues. This approach is not applicable for clinical samples due to the lack of paired references.[2] Reported success rate for the exact allocation of cells in brain tissue was 81%.[48]
DistMap
Mapping the transcriptome using the Distmap algorithm requires high-throughput single cell sequencing and an existing in situ hybridization atlas for the tissue of interest.[2][50] The Distmap algorithm generates a virtual 3D model of the tissue of interest using the transcriptomes of sequenced cells and said reference atlas.[2][50] The transcriptomes can be clustered into cell types using t-distributed stochastic neighbour embedding and mapped to the 3D model using virtual in situ hybridization.[50][51] Essentially, this algorithm takes data generated from single cells in a dissociated tissue and is able to map individual transcripts to where the cell type exists in the tissue using virtual in situ hybridization.
History
in situ hybridization was developed in the late 60's and saw major developments in the 80's (smFISH) and 10's (RNAscope, seqFISH, MERFISH and osmFISH) with the most recent additions being seqFISH+ and DNA microscopy.[15] Microdisecction techniques were first developed in the late 90's (Laser Capture Microdissection) and the most recent addition was in 2016 with the development of ProximID.[2] Spatial genomics/transcriptomics as a technique was invented and developed in 2000 by Michael Doyle (of Eolas), Maurice Pescitelli (of the University of Illinois at Chicago), Betsey Williams (of Harvard), and George Michaels (of George Mason University),[52][53][54] as part of the Visible Embryo Project[55] It was later expanded upon in 2016 by Jonas Frisén, Joakim Lundeberg, Patrik Ståhl and their colleagues in Stockholm, Sweden.[1] In 2019 at the Broad Institute, the labs of Fei Chen and Evan Macosko developed Slide-seq, which used barcoded oligos on beads.[44]
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. (July 2016). "Visualization and analysis of gene expression in tissue sections by spatial transcriptomics". Science. 353 (6294): 78–82. Bibcode:2016Sci...353...78S. doi:10.1126/science.aaf2403. PMID 27365449. S2CID 30942685.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 Asp M, Bergenstråhle J, Lundeberg J (October 2020). "Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration". BioEssays. 42 (10): e1900221. doi:10.1002/bies.201900221. PMID 32363691. S2CID 218492475.
- 1 2 "BioRender". BioRender. Retrieved 2021-02-26.
- 1 2 3 4 Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, et al. (November 1996). "Laser capture microdissection". Science. 274 (5289): 998–1001. Bibcode:1996Sci...274..998E. doi:10.1126/science.274.5289.998. PMID 8875945. S2CID 220105962.
- ↑ Simone NL, Bonner RF, Gillespie JW, Emmert-Buck MR, Liotta LA (July 1998). "Laser-capture microdissection: opening the microscopic frontier to molecular analysis". Trends in Genetics. 14 (7): 272–6. doi:10.1016/S0168-9525(98)01489-9. PMID 9676529.
- 1 2 3 4 Combs PA, Eisen MB (2013-08-12). "Sequencing mRNA from cryo-sliced Drosophila embryos to determine genome-wide spatial patterns of gene expression". PLOS ONE. 8 (8): e71820. arXiv:1302.4693. Bibcode:2013PLoSO...871820C. doi:10.1371/journal.pone.0071820. PMC 3741199. PMID 23951250.
- 1 2 3 4 5 6 Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, Fisher S, et al. (February 2014). "Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue". Nature Methods. 11 (2): 190–6. doi:10.1038/nmeth.2804. PMC 3964595. PMID 24412976.
- 1 2 3 4 Junker JP, Noël ES, Guryev V, Peterson KA, Shah G, Huisken J, et al. (October 2014). "Genome-wide RNA Tomography in the zebrafish embryo". Cell. 159 (3): 662–75. doi:10.1016/j.cell.2014.09.038. PMID 25417113. S2CID 2713635.
- 1 2 Nichterwitz S, Chen G, Aguila Benitez J, Yilmaz M, Storvall H, Cao M, Sandberg R, Deng Q, Hedlund E (July 2016). "Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling". Nature Communications. 7: 12139. Bibcode:2016NatCo...712139N. doi:10.1038/ncomms12139. PMC 4941116. PMID 27387371.
- ↑ Nichterwitz S, Aguila Benitez J, Hoogstraaten R, Deng Q, Hedlund E (2018). "LCM-Seq: A Method for Spatial Transcriptomic Profiling Using Laser Capture Microdissection Coupled with PolyA-Based RNA Sequencing". RNA Detection. Methods in Molecular Biology. Vol. 1649. pp. 95–110. doi:10.1007/978-1-4939-7213-5_6. ISBN 978-1-4939-7212-8. PMID 29130192.
- ↑ Nichterwitz S, Nijssen J, Storvall H, Schweingruber C, Comley LH, Allodi I, van der Lee M, Deng Q, Sandberg R, Hedlund E (August 2020). "LCM-seq reveals unique transcriptional adaptation mechanisms and protective pathways of resistant neurons in spinal muscular atrophy". Genome Research. 30 (8): 1083–1096. doi:10.1101/gr.265017.120. PMC 7462070. PMID 32820007.
- ↑ Pereira M, Birtele M, Shrigley S, Benitez JA, Hedlund E, Parmar M, Ottosson DR (September 2017). "Direct Reprogramming of Resident NG2 Glia into Neurons with Properties of Fast-Spiking Parvalbumin-Containing Interneurons". Stem Cell Reports. 9 (3): 742–751. doi:10.1016/j.stemcr.2017.07.023. PMC 5599255. PMID 28844658.
- ↑ Aguila J, Cheng S, Kee N, Cao M, Wang M, Deng Q, Hedlund E (2021). "Spatial RNA Sequencing Identifies Robust Markers of Vulnerable and Resistant Human Midbrain Dopamine Neurons and Their Expression in Parkinson's Disease". Frontiers in Molecular Neuroscience. 14: 699562. doi:10.3389/fnmol.2021.699562. PMC 8297217. PMID 34305528.
- ↑ Newton PT, Li L, Zhou B, Schweingruber C, Hovorakova M, Xie M, et al. (March 2019). "A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate". Nature. 567 (7747): 234–238. Bibcode:2019Natur.567..234N. doi:10.1038/s41586-019-0989-6. PMID 30814736. S2CID 71143703.
- 1 2 3 4 5 Chen J, Suo S, Tam PP, Han JJ, Peng G, Jing N (March 2017). "Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq". Nature Protocols. 12 (3): 566–580. doi:10.1038/nprot.2017.003. PMID 28207000. S2CID 3879096.
- 1 2 3 Peng G, Suo S, Chen J, Chen W, Liu C, Yu F, et al. (March 2016). "Spatial Transcriptome for the Molecular Annotation of Lineage Fates and Cell Identity in Mid-gastrula Mouse Embryo". Developmental Cell. 36 (6): 681–97. doi:10.1016/j.devcel.2016.02.020. PMID 27003939.
- 1 2 3 4 5 Medaglia C, Giladi A, Stoler-Barak L, De Giovanni M, Salame TM, Biram A, et al. (December 2017). "Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq". Science. 358 (6370): 1622–1626. Bibcode:2017Sci...358.1622M. doi:10.1126/science.aao4277. PMC 7234837. PMID 29217582.
- 1 2 3 4 Boisset JC, Vivié J, Grün D, Muraro MJ, Lyubimova A, van Oudenaarden A (July 2018). "Mapping the physical network of cellular interactions". Nature Methods. 15 (7): 547–553. doi:10.1038/s41592-018-0009-z. PMID 29786092. S2CID 29166537.
- 1 2 3 4 Chen X, Sun YC, Church GM, Lee JH, Zador AM (February 2018). "Efficient in situ barcode sequencing using padlock probe-based BaristaSeq". Nucleic Acids Research. 46 (4): e22. doi:10.1093/nar/gkx1206. PMC 5829746. PMID 29190363.
- 1 2 3 Femino AM, Fay FS, Fogarty K, Singer RH (April 1998). "Visualization of single RNA transcripts in situ". Science. 280 (5363): 585–90. Bibcode:1998Sci...280..585F. doi:10.1126/science.280.5363.585. PMID 9554849.
- ↑ Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (October 2008). "Imaging individual mRNA molecules using multiple singly labeled probes". Nature Methods. 5 (10): 877–9. doi:10.1038/nmeth.1253. PMC 3126653. PMID 18806792.
- 1 2 3 4 5 6 Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, et al. (January 2012). "RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues". The Journal of Molecular Diagnostics. 14 (1): 22–9. doi:10.1016/j.jmoldx.2011.08.002. PMC 3338343. PMID 22166544.
- ↑ De Biase, Davide; Prisco, Francesco; Piegari, Giuseppe; Ilsami, Arianna; d'Aquino, Ilaria; Baldassarre, Valeria; Zito Marino, Federica; Franco, Renato; Papparella, Serenella; Paciello, Orlando (16 February 2021). "RNAScope in situ Hybridization as a Novel Technique for the Assessment of c-KIT mRNA Expression in Canine Mast Cell Tumor". Frontiers in Veterinary Science. 8: 591961. doi:10.3389/fvets.2021.591961. PMC 7921150. PMID 33665215.
- 1 2 3 Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L (April 2014). "Single-cell in situ RNA profiling by sequential hybridization". Nature Methods. 11 (4): 360–1. doi:10.1038/nmeth.2892. PMC 4085791. PMID 24681720.
- ↑ Shah S, Lubeck E, Zhou W, Cai L (October 2016). "In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus". Neuron. 92 (2): 342–357. doi:10.1016/j.neuron.2016.10.001. PMC 5087994. PMID 27764670.
- ↑ Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X (April 2015). "RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells". Science. 348 (6233): aaa6090. doi:10.1126/science.aaa6090. PMC 4662681. PMID 25858977.
- 1 2 3 4 5 6 7 8 9 10 11 Zhuang X (January 2021). "Spatially resolved single-cell genomics and transcriptomics by imaging". Nature Methods. 18 (1): 18–22. doi:10.1038/s41592-020-01037-8. PMC 9805800. PMID 33408406. S2CID 230796841.
- 1 2 3 4 5 6 Shah S, Lubeck E, Schwarzkopf M, He TF, Greenbaum A, Sohn CH, et al. (August 2016). "Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing". Development. 143 (15): 2862–7. doi:10.1242/dev.138560. PMC 5004914. PMID 27342713.
- 1 2 3 4 Codeluppi S, Borm LE, Zeisel A, La Manno G, van Lunteren JA, Svensson CI, Linnarsson S (November 2018). "Spatial organization of the somatosensory cortex revealed by osmFISH". Nature Methods. 15 (11): 932–935. doi:10.1038/s41592-018-0175-z. PMID 30377364. S2CID 53114385.
- 1 2 Chen F, Wassie AT, Cote AJ, Sinha A, Alon S, Asano S, et al. (August 2016). "Nanoscale imaging of RNA with expansion microscopy". Nature Methods. 13 (8): 679–684. doi:10.1038/nmeth.3899. PMC 4965288. PMID 27376770.
- ↑ Wang Y, Eddison M, Fleishman G, Weigert M, Xu S, Wang T, et al. (December 2021). "EASI-FISH for thick tissue defines lateral hypothalamus spatio-molecular organization". Cell. 184 (26): 6361–6377.e24. doi:10.1016/j.cell.2021.11.024. PMID 34875226. S2CID 244906189.
- ↑ Wang Y, Eddison M, Fleishman G, Weigert M, Xu S, Wang T, et al. (December 2021). "EASI-FISH for thick tissue defines lateral hypothalamus spatio-molecular organization". Cell. 184 (26): 6361–6377.e24. doi:10.1016/j.cell.2021.11.024. PMID 34875226. S2CID 244906189.
- 1 2 3 Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. (April 2019). "Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH". Nature. 568 (7751): 235–239. Bibcode:2019Natur.568..235E. doi:10.1038/s41586-019-1049-y. PMC 6544023. PMID 30911168.
- 1 2 3 4 5 6 Weinstein JA, Regev A, Zhang F (June 2019). "DNA Microscopy: Optics-free Spatio-genetic Imaging by a Stand-Alone Chemical Reaction". Cell. 178 (1): 229–241.e16. doi:10.1016/j.cell.2019.05.019. PMC 6697087. PMID 31230717.
- ↑ Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wählby C, Nilsson M (September 2013). "In situ sequencing for RNA analysis in preserved tissue and cells". Nature Methods. 10 (9): 857–60. doi:10.1038/nmeth.2563. PMID 23852452. S2CID 205421785.
- ↑ Szemes M, Bonants P, de Weerdt M, Baner J, Landegren U, Schoen CD (April 2005). "Diagnostic application of padlock probes--multiplex detection of plant pathogens using universal microarrays". Nucleic Acids Research. 33 (8): e70. doi:10.1093/nar/gni069. PMC 1087788. PMID 15860767.
- ↑ Ali MM, Li F, Zhang Z, Zhang K, Kang DK, Ankrum JA, et al. (May 2014). "Rolling circle amplification: a versatile tool for chemical biology, materials science and medicine". Chemical Society Reviews. 43 (10): 3324–41. doi:10.1039/c3cs60439j. PMID 24643375. S2CID 24839827.
- ↑ Churko JM, Mantalas GL, Snyder MP, Wu JC (June 2013). "Overview of high throughput sequencing technologies to elucidate molecular pathways in cardiovascular diseases". Circulation Research. 112 (12): 1613–23. doi:10.1161/CIRCRESAHA.113.300939. PMC 3831009. PMID 23743227.
- ↑ Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. (March 2014). "Highly multiplexed subcellular RNA sequencing in situ". Science. 343 (6177): 1360–3. Bibcode:2014Sci...343.1360L. doi:10.1126/science.1250212. PMC 4140943. PMID 24578530.
- ↑ Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. (July 2018). "Three-dimensional intact-tissue sequencing of single-cell transcriptional states". Science. 361 (6400): eaat5691. doi:10.1126/science.aat5691. PMC 6339868. PMID 29930089.
- ↑ "STOmics Stereo-seq Spatial Transcriptomics Technology".
- ↑ https://www.cell.com/cell/fulltext/S0092-8674(22)00399-3?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0092867422003993%3Fshowall%3Dtrue
- 1 2 Zollinger DR, Lingle SE, Sorg K, Beechem JM, Merritt CR (2020). "GeoMx™ RNA Assay: High Multiplex, Digital, Spatial Analysis of RNA in FFPE Tissue". In Nielsen BS, Jones J (eds.). In Situ Hybridization Protocols. Methods in Molecular Biology. Vol. 2148. Clifton, N.J. pp. 331–345. doi:10.1007/978-1-0716-0623-0_21. ISBN 978-1-0716-0623-0. PMID 32394392. S2CID 218599146.
{{cite book}}: CS1 maint: location missing publisher (link) - 1 2 3 4 5 Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. (March 2019). "Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution". Science. 363 (6434): 1463–1467. Bibcode:2019Sci...363.1463R. doi:10.1126/science.aaw1219. PMC 6927209. PMID 30923225.
- 1 2 3 4 Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, et al. (July 2019). "Atlas of Subcellular RNA Localization Revealed by APEX-Seq". Cell. 178 (2): 473–490.e26. doi:10.1016/j.cell.2019.05.027. PMC 6786773. PMID 31230715.
- 1 2 3 4 Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, et al. (October 2019). "High-definition spatial transcriptomics for in situ tissue profiling". Nature Methods. 16 (10): 987–990. doi:10.1038/s41592-019-0548-y. hdl:1721.1/126032. PMC 6765407. PMID 31501547.
- ↑ "Spatial Gene Expression".
- 1 2 Achim K, Pettit JB, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, Marioni JC (May 2015). "High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin". Nature Biotechnology. 33 (5): 503–9. doi:10.1038/nbt.3209. PMID 25867922. S2CID 19535470.
- ↑ Satija R, Farrell JA, Gennert D, Schier AF, Regev A (May 2015). "Spatial reconstruction of single-cell gene expression data". Nature Biotechnology. 33 (5): 495–502. doi:10.1038/nbt.3192. PMC 4430369. PMID 25867923.
- 1 2 3 Karaiskos N, Wahle P, Alles J, Boltengagen A, Ayoub S, Kipar C, et al. (October 2017). "The Drosophila embryo at single-cell transcriptome resolution". Science. 358 (6360): 194–199. Bibcode:2017Sci...358..194K. doi:10.1126/science.aan3235. PMID 28860209. S2CID 206659563.
- ↑ Kobak D, Berens P (November 2019). "The art of using t-SNE for single-cell transcriptomics". Nature Communications. 10 (1): 5416. Bibcode:2019NatCo..10.5416K. doi:10.1038/s41467-019-13056-x. PMC 6882829. PMID 31780648.
- ↑ US 7613571, Doyle, Michael D.; Pescitelli, Jr., Maurice J. & Williams, Betsey S. et al., "Method and system for the multidimensional morphological reconstruction of genome expression activity", published 2009-11-03
- ↑ US 7894997, Doyle, Michael D.; Pescitelli, Jr., Maurice J. & Williams, Betsey S. et al., "Multidimensional morphological reconstruction of genome expression activity", published 2011-02-22
- ↑ US 10011864, Doyle, Michael D.; Pescitelli, Jr., Maurice J. & Williams, Betsey S. et al., "Multidimensional microdissection and morphological reconstruction of genomic or proteomic expression activity", published 2018-07-03, assigned to Eolas Technologies Inc.
- ↑ Doyle MD, Noe A, Michaels GS (2000). Oliver WR (ed.). "The Visible Embryo Project: A Platform for Spatial Genomics". Proceedings of SPIE. 28th AIPR Workshop: 3D Visualization for Data Exploration and Decision Making. 3905: 248. Bibcode:2000SPIE.3905..248D. doi:10.1117/12.384880. S2CID 85027703.