The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.
The early stages of transcriptome annotations began with cDNA libraries published in the 1980s. Subsequently, the advent of high-throughput technology led to faster and more efficient ways of obtaining data about the transcriptome. Two biological techniques are used to study the transcriptome, namely DNA microarray, a hybridization-based technique and RNA-seq, a sequence-based approach.[1] RNA-seq is the preferred method and has been the dominant transcriptomics technique since the 2010s. Single-cell transcriptomics allows tracking of transcript changes over time within individual cells.
Data obtained from the transcriptome is used in research to gain insight into processes such as cellular differentiation, carcinogenesis, transcription regulation and biomarker discovery among others. Transcriptome-obtained data also finds applications in establishing phylogenetic relationships during the process of evolution and in in vitro fertilization. The transcriptome is closely related to other -ome based biological fields of study; it is complementary to the proteome and the metabolome and encompasses the translatome, exome, meiome and thanatotranscriptome which can be seen as ome fields studying specific types of RNA transcripts. There are quantifiable and conserved relationships between the Transcriptome and other -omes, and Transcriptomics data can be used effectively to predict other molecular species, such as metabolites.[2] There are numerous publicly available transcriptome databases.
Etymology and history
The word transcriptome is a portmanteau of the words transcript and genome. It appeared along with other neologisms formed using the suffixes -ome and -omics to denote all studies conducted on a genome-wide scale in the fields of life sciences and technology. As such, transcriptome and transcriptomics were one of the first words to emerge along with genome and proteome.[3] The first study to present a case of a collection of a cDNA library for silk moth mRNA was published in 1979.[4] The first seminal study to mention and investigate the transcriptome of an organism was published in 1997 and it described 60,633 transcripts expressed in S. cerevisiae using serial analysis of gene expression (SAGE).[5] With the rise of high-throughput technologies and bioinformatics and the subsequent increased computational power, it became increasingly efficient and easy to characterize and analyze enormous amount of data.[3] Attempts to characterize the transcriptome became more prominent with the advent of automated DNA sequencing during the 1980s.[6] During the 1990s, expressed sequence tag sequencing was used to identify genes and their fragments.[7] This was followed by techniques such as serial analysis of gene expression (SAGE), cap analysis of gene expression (CAGE), and massively parallel signature sequencing (MPSS).
Transcription
The transcriptome encompasses all the ribonucleic acid (RNA) transcripts present in a given organism or experimental sample.[8] RNA is the main carrier of genetic information that is responsible for the process of converting DNA into an organism's phenotype. A gene can give rise to a single-stranded messenger RNA (mRNA) through a molecular process known as transcription; this mRNA is complementary to the strand of DNA it originated from.[6] The enzyme RNA polymerase II attaches to the template DNA strand and catalyzes the addition of ribonucleotides to the 3' end of the growing sequence of the mRNA transcript.[9]
In order to initiate its function, RNA polymerase II needs to recognize a promoter sequence, located upstream (5') of the gene. In eukaryotes, this process is mediated by transcription factors, most notably Transcription factor II D (TFIID) which recognizes the TATA box and aids in the positioning of RNA polymerase at the appropriate start site. To finish the production of the RNA transcript, termination takes place usually several hundred nuclecotides away from the termination sequence and cleavage takes place.[9] This process occurs in the nucleus of a cell along with RNA processing by which mRNA molecules are capped, spliced and polyadenylated to increase their stability before being subsequently taken to the cytoplasm. The mRNA gives rise to proteins through the process of translation that takes place in ribosomes.
Types of RNA transcripts
Almost all functional transcripts are derived from known genes. The only exceptions are a small number of transcripts that might play a direct role in regulating gene expression near the prompters of known genes. (See Enhancer RNA.)
Gene occupy most of prokaryotic genomes so most of their genomes are transcribed. Many eukaryotic genomes are very large and known genes may take up only a fraction of the genome. In mammals, for example, known genes only account for 40-50% of the genome.[10] Nevertheless, identified transcripts often map to a much larger fraction of the genome suggesting that the transcriptome contains spurious transcripts that do not come from genes. Some of these transcripipts are known to be non-functional because they map to transcribed pseudogenes or degenerative transposons and viruses. Others map to unidentified regions of the genome that may be junk DNA.
Spurious transcription is very common in eukaryotes, especially those with large genomes that might contain a lot of junk DNA.[11][12][13][14] Some scientists claim that if a transcript has not been assigned to a known gene then the default assumption must be that it is junk RNA until it has been shown to be functional.[11][15] This would mean that much of the transcriptome in species with large genomes is probably junk RNA. (See Non-coding RNA)
The transcriptome includes the transcripts of protein-coding genes (mRNA plus introns) as well as the transcripts of non-coding genes (functional RNAs plus introns).
- Ribosomal RNA/rRNA: Usually the most abundant RNA in the transcriptome.
- Long non-coding RNA/lncRNA: Non-coding RNA transcripts that are more than 200 nucleotides long. Members of this group comprise the largest fraction of the non-coding transcriptome other than introns. It is not known how many of these transcripts are functional and how many are junk RNA.
- transfer RNA/tRNA
- micro RNA/miRNA: 19-24 nucleotides (nt) long. Micro RNAs up- or downregulate expression levels of mRNAs by the process of RNA interference at the post-transcriptional level.[3]
- small interfering RNA/siRNA: 20-24 nt
- small nucleolar RNA/snoRNA
- Piwi-interacting RNA/piRNA: 24-31 nt. They interact with Piwi proteins of the Argonaute family and have a function in targeting and cleaving transposons.[16]
- enhancer RNA/eRNA:[3]
Scope of study
In the human genome, all genes get transcribed into RNA because that's how the molecular gene is defined. (See Gene.) The transcriptome consists of coding regions of mRNA plus non-coding UTRs, introns, non-coding RNAs, and spurious non-functional transcripts.
Several factors render the content of the transcriptome difficult to establish. These include alternative splicing, RNA editing and alternative transcription among others.[17] Additionally, transcriptome techniques are capable of capturing transcription occurring in a sample at a specific time point, although the content of the transcriptome can change during differentiation.[6] The main aims of transcriptomics are the following: "catalogue all species of transcript, including mRNAs, non-coding RNAs and small RNAs; to determine the transcriptional structure of genes, in terms of their start sites, 5′ and 3′ ends, splicing patterns and other post-transcriptional modifications; and to quantify the changing expression levels of each transcript during development and under different conditions".[1]
The term can be applied to the total set of transcripts in a given organism, or to the specific subset of transcripts present in a particular cell type. Unlike the genome, which is roughly fixed for a given cell line (excluding mutations), the transcriptome can vary with external environmental conditions. Because it includes all mRNA transcripts in the cell, the transcriptome reflects the genes that are being actively expressed at any given time, with the exception of mRNA degradation phenomena such as transcriptional attenuation. The study of transcriptomics, (which includes expression profiling, splice variant analysis etc.), examines the expression level of RNAs in a given cell population, often focusing on mRNA, but sometimes including others such as tRNAs and sRNAs.
Methods of construction
Transcriptomics is the quantitative science that encompasses the assignment of a list of strings ("reads") to the object ("transcripts" in the genome). To calculate the expression strength, the density of reads corresponding to each object is counted.[18] Initially, transcriptomes were analyzed and studied using expressed sequence tags libraries and serial and cap analysis of gene expression (SAGE).
Currently, the two main transcriptomics techniques include DNA microarrays and RNA-Seq. Both techniques require RNA isolation through RNA extraction techniques, followed by its separation from other cellular components and enrichment of mRNA.[19][20]
There are two general methods of inferring transcriptome sequences. One approach maps sequence reads onto a reference genome, either of the organism itself (whose transcriptome is being studied) or of a closely related species. The other approach, de novo transcriptome assembly, uses software to infer transcripts directly from short sequence reads and is used in organisms with genomes that are not sequenced.[21]
DNA microarrays

The first transcriptome studies were based on microarray techniques (also known as DNA chips). Microarrays consist of thin glass layers with spots on which oligonucleotides, known as "probes" are arrayed; each spot contains a known DNA sequence.[22]
When performing microarray analyses, mRNA is collected from a control and an experimental sample, the latter usually representative of a disease. The RNA of interest is converted to cDNA to increase its stability and marked with fluorophores of two colors, usually green and red, for the two groups. The cDNA is spread onto the surface of the microarray where it hybridizes with oligonucleotides on the chip and a laser is used to scan. The fluorescence intensity on each spot of the microarray corresponds to the level of gene expression and based on the color of the fluorophores selected, it can be determined which of the samples exhibits higher levels of the mRNA of interest.[7]
One microarray usually contains enough oligonucleotides to represent all known genes; however, data obtained using microarrays does not provide information about unknown genes. During the 2010s, microarrays were almost completely replaced by next-generation techniques that are based on DNA sequencing.
RNA sequencing
RNA sequencing is a next-generation sequencing technology; as such it requires only a small amount of RNA and no previous knowledge of the genome.[3] It allows for both qualitative and quantitative analysis of RNA transcripts, the former allowing discovery of new transcripts and the latter a measure of relative quantities for transcripts in a sample.[16]
The three main steps of sequencing transcriptomes of any biological samples include RNA purification, the synthesis of an RNA or cDNA library and sequencing the library.[16] The RNA purification process is different for short and long RNAs.[16] This step is usually followed by an assessment of RNA quality, with the purpose of avoiding contaminants such as DNA or technical contaminants related to sample processing. RNA quality is measured using UV spectrometry with an absorbance peak of 260 nm.[23] RNA integrity can also be analyzed quantitatively comparing the ratio and intensity of 28S RNA to 18S RNA reported in the RNA Integrity Number (RIN) score.[23] Since mRNA is the species of interest and it represents only 3% of its total content, the RNA sample should be treated to remove rRNA and tRNA and tissue-specific RNA transcripts.[23]
The step of library preparation with the aim of producing short cDNA fragments, begins with RNA fragmentation to transcripts in length between 50 and 300 base pairs. Fragmentation can be enzymatic (RNA endonucleases), chemical (trismagnesium salt buffer, chemical hydrolysis) or mechanical (sonication, nebulisation).[24] Reverse transcription is used to convert the RNA templates into cDNA and three priming methods can be used to achieve it, including oligo-DT, using random primers or ligating special adaptor oligos.
Single-cell transcriptomics
Transcription can also be studied at the level of individual cells by single-cell transcriptomics. Single-cell RNA sequencing (scRNA-seq) is a recently developed technique that allows the analysis of the transcriptome of single cells, including bacteria.[25] With single-cell transcriptomics, subpopulations of cell types that constitute the tissue of interest are also taken into consideration.[26] This approach allows to identify whether changes in experimental samples are due to phenotypic cellular changes as opposed to proliferation, with which a specific cell type might be overexpressed in the sample.[27] Additionally, when assessing cellular progression through differentiation, average expression profiles are only able to order cells by time rather than their stage of development and are consequently unable to show trends in gene expression levels specific to certain stages.[28] Single-cell trarnscriptomic techniques have been used to characterize rare cell populations such as circulating tumor cells, cancer stem cells in solid tumors, and embryonic stem cells (ESCs) in mammalian blastocysts.[29]
Although there are no standardized techniques for single-cell transcriptomics, several steps need to be undertaken. The first step includes cell isolation, which can be performed using low- and high-throughput techniques. This is followed by a qPCR step and then single-cell RNAseq where the RNA of interest is converted into cDNA. Newer developments in single-cell transcriptomics allow for tissue and sub-cellular localization preservation through cryo-sectioning thin slices of tissues and sequencing the transcriptome in each slice. Another technique allows the visualization of single transcripts under a microscope while preserving the spatial information of each individual cell where they are expressed.[29]
Analysis
A number of organism-specific transcriptome databases have been constructed and annotated to aid in the identification of genes that are differentially expressed in distinct cell populations.
RNA-seq is emerging (2013) as the method of choice for measuring transcriptomes of organisms, though the older technique of DNA microarrays is still used.[1] RNA-seq measures the transcription of a specific gene by converting long RNAs into a library of cDNA fragments. The cDNA fragments are then sequenced using high-throughput sequencing technology and aligned to a reference genome or transcriptome which is then used to create an expression profile of the genes.[1]
Applications
Mammals
The transcriptomes of stem cells and cancer cells are of particular interest to researchers who seek to understand the processes of cellular differentiation and carcinogenesis. A pipeline using RNA-seq or gene array data can be used to track genetic changes occurring in stem and precursor cells and requires at least three independent gene expression data from the former cell type and mature cells.[30]
Analysis of the transcriptomes of human oocytes and embryos is used to understand the molecular mechanisms and signaling pathways controlling early embryonic development, and could theoretically be a powerful tool in making proper embryo selection in in vitro fertilisation. Analyses of the transcriptome content of the placenta in the first-trimester of pregnancy in in vitro fertilization and embryo transfer (IVT-ET) revealed differences in genetic expression which are associated with higher frequency of adverse perinatal outcomes. Such insight can be used to optimize the practice.[31] Transcriptome analyses can also be used to optimize cryopreservation of oocytes, by lowering injuries associated with the process.[32]
Transcriptomics is an emerging and continually growing field in biomarker discovery for use in assessing the safety of drugs or chemical risk assessment.[33]
Transcriptomes may also be used to infer phylogenetic relationships among individuals or to detect evolutionary patterns of transcriptome conservation.[34]
Transcriptome analyses were used to discover the incidence of antisense transcription, their role in gene expression through interaction with surrounding genes and their abundance in different chromosomes.[35] RNA-seq was also used to show how RNA isoforms, transcripts stemming from the same gene but with different structures, can produce complex phenotypes from limited genomes.[21]
Plants
Transcriptome analysis have been used to study the evolution and diversification process of plant species. In 2014, the 1000 Plant Genomes Project was completed in which the transcriptomes of 1,124 plant species from the families viridiplantae, glaucophyta and rhodophyta were sequenced. The protein coding sequences were subsequently compared to infer phylogenetic relationships between plants and to characterize the time of their diversification in the process of evolution.[36] Transcriptome studies have been used to characterize and quantify gene expression in mature pollen. Genes involved in cell wall metabolism and cytoskeleton were found to be overexpressed. Transcriptome approaches also allowed to track changes in gene expression through different developmental stages of pollen, ranging from microspore to mature pollen grains; additionally such stages could be compared across species of different plants including Arabidopsis, rice and tobacco.[37]
Relation to other ome fields
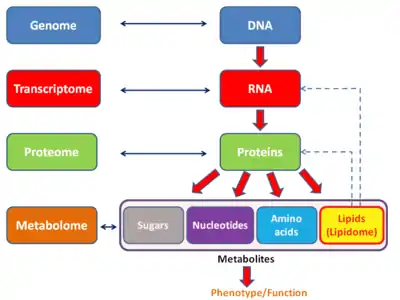
Similar to other -ome based technologies, analysis of the transcriptome allows for an unbiased approach when validating hypotheses experimentally. This approach also allows for the discovery of novel mediators in signaling pathways.[18] As with other -omics based technologies, the transcriptome can be analyzed within the scope of a multiomics approach. It is complementary to metabolomics but contrary to proteomics, a direct association between a transcript and metabolite cannot be established.
There are several -ome fields that can be seen as subcategories of the transcriptome. The exome differs from the transcriptome in that it includes only those RNA molecules found in a specified cell population, and usually includes the amount or concentration of each RNA molecule in addition to the molecular identities. Additionally, the transcritpome also differs from the translatome, which is the set of RNAs undergoing translation.
The term meiome is used in functional genomics to describe the meiotic transcriptome or the set of RNA transcripts produced during the process of meiosis.[38] Meiosis is a key feature of sexually reproducing eukaryotes, and involves the pairing of homologous chromosome, synapse and recombination. Since meiosis in most organisms occurs in a short time period, meiotic transcript profiling is difficult due to the challenge of isolation (or enrichment) of meiotic cells (meiocytes). As with transcriptome analyses, the meiome can be studied at a whole-genome level using large-scale transcriptomic techniques.[39] The meiome has been well-characterized in mammal and yeast systems and somewhat less extensively characterized in plants.[40]
The thanatotranscriptome consists of all RNA transcripts that continue to be expressed or that start getting re-expressed in internal organs of a dead body 24–48 hours following death. Some genes include those that are inhibited after fetal development. If the thanatotranscriptome is related to the process of programmed cell death (apoptosis), it can be referred to as the apoptotic thanatotranscriptome. Analyses of the thanatotranscriptome are used in forensic medicine.[41]
eQTL mapping can be used to complement genomics with transcriptomics; genetic variants at DNA level and gene expression measures at RNA level.[42]
Relation to proteome
The transcriptome can be seen as a subset of the proteome, that is, the entire set of proteins expressed by a genome.
However, the analysis of relative mRNA expression levels can be complicated by the fact that relatively small changes in mRNA expression can produce large changes in the total amount of the corresponding protein present in the cell. One analysis method, known as gene set enrichment analysis, identifies coregulated gene networks rather than individual genes that are up- or down-regulated in different cell populations.
Although microarray studies can reveal the relative amounts of different mRNAs in the cell, levels of mRNA are not directly proportional to the expression level of the proteins they code for.[43] The number of protein molecules synthesized using a given mRNA molecule as a template is highly dependent on translation-initiation features of the mRNA sequence; in particular, the ability of the translation initiation sequence is a key determinant in the recruiting of ribosomes for protein translation.
Transcriptome databases
See also
Notes
- 1 2 3 4 Wang, Zhong; Gerstein, Mark; Snyder, Michael (January 2009). "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
- ↑ Cavicchioli, Maria Vittoria; Santorsola, Mariangela; Balboni, Nicola; Mercatelli, Daniele; Giorgi, Federico Manuel (January 2022). "Prediction of Metabolic Profiles from Transcriptomics Data in Human Cancer Cell Lines". International Journal of Molecular Sciences. 23 (7): 3867. doi:10.3390/ijms23073867. ISSN 1422-0067. PMC 8998886. PMID 35409231.
- 1 2 3 4 5 Jiménez-Chillarón, Josep C.; Díaz, Rubén; Ramón-Krauel, Marta (2014). "Chapter 4 - Omics Tools for the Genome-Wide Analysis of Methylation and Histone Modifications". Comprehensive Analytical Chemistry. 64: 81–110. doi:10.1016/B978-0-444-62651-6.00004-0. ISBN 9780444626516. Retrieved 25 April 2020.
- ↑ GK, Sim; FC, Kafatos; CW, Jones; MD, Koehler; A, Efstratiadis; T., Maniatis (December 1979). "Use of a cDNA library for studies on evolution and developmental expression of the chorion multigene families". Cell. 8 (4): 1303–16. doi:10.1016/0092-8674(79)90241-1. PMID 519770.
- ↑ E Velculescu, Victor; Zhang, Lin; Zhou, Wei; Vogelstein, Jacob; A Basrai, Munira; E Bassett Jr., Douglas; Hieter, Phil; Vogelstein, Bert; W Kinzler, Kenneth (1997). "Characterization of the Yeast Transcriptome". Cell. 2 (88): 243–51. doi:10.1016/S0092-8674(00)81845-0. PMID 9008165. S2CID 11430660.
- 1 2 3 Peralta, Mihaela (2012). "The Human Transcriptome: An Unfinished Story". Genes. 3 (3): 344–360. doi:10.3390/genes3030344. PMC 3422666. PMID 22916334.
- 1 2 Govindarajan, Rajeshwar; Duraiyan, Jeyapradha; Kaliyappan, Karunakaran; Palanisamy, Murugesan (2012). "Microarray and its applications". Journal of Pharmacy and Bioallied Sciences. 4 (6): S310-2. doi:10.4103/0975-7406.100283. PMC 3467903. PMID 23066278.
- ↑ Brown, TA (2018). "Chapter 12: Transcriptomics". Genomes 4. New York, NY, USA: Garland Science. ISBN 9780815345084.
- 1 2 Clancy, Suzanne (2008). "DNA Transcription". Nature Education. 1 (11): 41.
- ↑ Francis WR, Wörheide G (June 2017). "Similar Ratios of Introns to Intergenic Sequence across Animal Genomes". Genome Biology and Evolution. 9 (6): 1582–1598. doi:10.1093/gbe/evx103. PMC 5534336. PMID 28633296.
- 1 2 van Bakel H, Nislow C, Blencowe BJ, and Hughes TR (2011). "Response to "the reality of pervasive transcription". PLOS Biology. 9 (7): e1001102. doi:10.1371/journal.pbio.1001102. PMC 3134445. S2CID 15680321.
- ↑ Jensen TH, Jacquier A, and Libri D (2013). "Dealing with pervasive transcription". Molecular Cell. 52 (4): 473–484. doi:10.1016/j.molcel.2013.10.032. PMID 24267449.
- ↑ Sverdlov, Eugene (2017). "Transcribed Junk Remains Junk If It Does Not Acquire A Selected Function in Evolution". BioEssays. 39 (12): 1700164. doi:10.1002/bies.201700164. PMID 29071727. S2CID 35346807.
- ↑ Wade JT, and Grainger DC (2018). "Spurious transcription and its impact on cell function". Transcription. 9 (3): 182–189. doi:10.1080/21541264.2017.1381794. PMC 5927700. PMID 28980880.
- ↑ Palazzo AF, and Lee ES (2015). "Non-coding RNA: what is functional and what is junk?". Frontiers in Genetics. 6: 2. doi:10.3389/fgene.2015.00002. PMC 4306305. PMID 25674102.
- 1 2 3 4 Cellerino & Sanguanini 2018, p. 12
- ↑ U. Adams, Jill (2008). "Transcriptome: Connecting the Genome to Gene Function". Nature Education. 1 (1): 195.
- 1 2 Cellerino & Sanguanini 2018, p. preface
- ↑ Bryant S, Manning DL (1998). "Isolation of messenger RNA". RNA Isolation and Characterization Protocols. Methods in Molecular Biology. Vol. 86. pp. 61–4. doi:10.1385/0-89603-494-1:61. ISBN 978-0-89603-494-5. PMID 9664454.
- ↑ Chomczynski P, Sacchi N (April 1987). "Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction". Analytical Biochemistry. 162 (1): 156–9. doi:10.1016/0003-2697(87)90021-2. PMID 2440339.
- 1 2 Tachibana, Chris (31 July 2015). "Transcriptomics today: Microarrays, RNA-seq, and more". Science Magazine. 349 (6247): 544. Bibcode:2015Sci...349..544T. Retrieved 2 May 2020.
- ↑ Schena, M.; Shalon, D.; Davis, R. W.; Brown, P. O. (20 October 1995). "Quantitative monitoring of gene expression patterns with a complementary DNA microarray". Science. New York, N.Y. 270 (5235): 467–470. Bibcode:1995Sci...270..467S. doi:10.1126/science.270.5235.467. ISSN 0036-8075. PMID 7569999. S2CID 6720459.
- 1 2 3 Cellerino & Sanguanini 2018, p. 13
- ↑ Cellerino & Sanguanini 2018, p. 18
- ↑ Toledo-Arana A, Lasa I (March 2020). "Advances in bacterial transcriptome understanding: From overlapping transcription to the excludon concept". Mol Microbiol. 113 (3): 593–602. doi:10.1111/mmi.14456. PMC 7154746. PMID 32185833.
- ↑ Kanter, Itamar; Kalisky, Tomer (10 March 2015). "Single Cell Transcriptomics: Methods and Applications". Frontiers in Oncology. 5: 53. doi:10.3389/fonc.2015.00053. ISSN 2234-943X. PMC 4354386. PMID 25806353.
- ↑ Stegle, Oliver; A. Teichmann, Sarah; C. Marioni, John (2015). "Computational and analytical challenges in single-cell transcriptomics". Nature Reviews Genetics. 16 (3): 133–45. doi:10.1038/nrg3833. PMID 25628217. S2CID 205486032.
- ↑ Trapnell, Cole (1 October 2015). "Defining cell types and states with single-cell genomics". Genome Research. 25 (10): 1491–1498. doi:10.1101/gr.190595.115. ISSN 1088-9051. PMC 4579334. PMID 26430159.
- 1 2 Kanter, Itamar; Kalisky, Tomer (2015). "Single Cell Transcriptomics: Methods and Applications". Frontiers in Oncology. 5 (13): 53. doi:10.3389/fonc.2015.00053. PMC 4354386. PMID 25806353.
- ↑ Godoy, Patricio; Schmidt-Heck, Wolfgang; Hellwig, Birte; Nell, Patrick; Feuerborn, David; Rahnenführer, Jörg; Kattler, Kathrin; Walter, Jörn; Blüthgen, Nils; G. Hengstler, Jan (5 July 2018). "Assessment of stem cell differentiation based on genome-wide expression profiles". Philosophical Transactions of the Royal Society B. 373 (1750): 20170221. doi:10.1098/rstb.2017.0221. PMC 5974444. PMID 29786556.
- ↑ Zhao, L; Zheng, X; Liu, J; Zheng, R; Yang, R; Wang, Y; Sun, L (1 July 2019). "The placental transcriptome of the first-trimester placenta is affected by in vitro fertilization and embryo transfer". Reproductive Biology and Endocrinology. 17 (1): 50. doi:10.1186/s12958-019-0494-7. PMC 6604150. PMID 31262321.
- ↑ Eroglu, Binnur; A. Szurek, Edyta; Schall, Peter; E. Latham, Keith; Eroglu, Ali (6 April 2020). "Probing lasting cryoinjuries to oocyte-embryo transcriptome". PLOS ONE. 15 (4): e0231108. Bibcode:2020PLoSO..1531108E. doi:10.1371/journal.pone.0231108. PMC 7135251. PMID 32251418.
- ↑ Szabo, David (2014). "Transcriptomic biomarkers in safety and risk assessment of chemicals". Transcriptomic biomarkers in safety and risk assessment of chemicals. In Ramesh Gupta, editors:Gupta - Biomarkers in Toxicology, Oxford:Academic Press. pp. 1033–1038. doi:10.1016/B978-0-12-404630-6.00062-2. ISBN 978-0-12-404630-6. S2CID 89396307.
- ↑ Drost, Hajk-Georg; Gabel, Alexander; Grosse, Ivo; Quint, Marcel; Grosse, Ivo (2018-05-01). "myTAI: evolutionary transcriptomics with R". Bioinformatics. 34 (9): 1589–1590. doi:10.1093/molbev/msv012. ISSN 0737-4038. PMC 5925770. PMID 29309527.
- ↑ S, Katayama; et al. (2005). "Antisense Transcription in the Mammalian Transcriptome". Science. 309 (5740): 1564–6. Bibcode:2005Sci...309.1564R. doi:10.1126/science.1112009. PMID 16141073. S2CID 34559885.
- ↑ One Thousand Plant Transcriptomes Initiative (23 October 2019). "One thousand plant transcriptomes and the phylogenomics of green plants". Nature. 574 (7780): 679–685. doi:10.1038/s41586-019-1693-2. PMC 6872490. PMID 31645766.
- ↑ Rutley, Nicholas; Twell, David (12 March 2015). "A decade of pollen transcriptomics". Plant Reproduction. 28 (2): 73–89. doi:10.1007/s00497-015-0261-7. PMC 4432081. PMID 25761645.
- ↑ Crismani, Wayne; Baumann, Ute; Sutton, Tim; Shirley, Neil; Webster, Tracie; Spangenberg, German; Langridge, Peter; A Able, Jason (2006). "Microarray expression analysis of meiosis and microsporogenesis in hexaploid bread wheat". BMC Genomics. 7 (267): 267. doi:10.1186/1471-2164-7-267. PMC 1647286. PMID 17052357.
- ↑ D. Bovill, William; Deveshwar, Priyanka; Kapoor, Sanjay; A. Able, Jason (2009). "Whole genome approaches to identify early meiotic gene candidates in cereals". Functional & Integrative Genomics. 9 (2): 219–29. doi:10.1007/s10142-008-0097-4. PMID 18836753. S2CID 22854431.
- ↑ Deveshwar, Priyanka; D Bovill, William; Sharma, Rita; A Able, Jason; Kapoor, Sanjay (9 May 2011). "Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice". BMC Plant Biology. 11 (78): 78. doi:10.1186/1471-2229-11-78. PMC 3112077. PMID 21554676.
- ↑ Javan, G. T.; Can, I.; Finley, S. J.; Soni, S (2015). "The apoptotic thanatotranscriptome associated with the liver of cadavers". Forensic Science, Medicine, and Pathology. 11 (4): 509–516. doi:10.1007/s12024-015-9704-6. PMID 26318598. S2CID 21583165.
- ↑ Manzoni, Claudia; A Kia, Demis; Vandrovcova, Jana; Hardy, John; W Wood, Nicholas; A Lewis, Patrick; Ferrari, Raffaele (March 2018). "Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences". Briefings in Bioinformatics. 19 (2): 286–302. doi:10.1093/bib/bbw114. PMC 6018996. PMID 27881428.
- ↑ Schwanhäusser, Björn; et al. (May 2011). "Global quantification of mammalian gene expression control" (PDF). Nature. 473 (7347): 337–342. Bibcode:2011Natur.473..337S. doi:10.1038/nature10098. PMID 21593866. S2CID 205224972.
References
- Cellerino, A; Sanguanini, M (2018), Transcriptome Analysis: Introduction and Examples from the Neurosciences, doi:10.1007/978-88-7642-642-1, ISBN 978-88-7642-641-4
Further reading
- ^ Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545-50.
- ^ Laule O, Hirsch-Hoffmann M, Hruz T, Gruissem W, and P Zimmermann. (2006) Web-based analysis of the mouse transcriptome using Genevestigator. BMC Bioinformatics 7:311
- ^ Assou, S.; Boumela, I.; Haouzi, D.; Anahory, T.; Dechaud, H.; De Vos, J.; Hamamah, S. (2010). "Dynamic changes in gene expression during human early embryo development: From fundamental aspects to clinical applications". Human Reproduction Update. 17 (2): 272–290. doi:10.1093/humupd/dmq036. PMC 3189516. PMID 20716614.
- ^ Ogorodnikov, A; Kargapolova, Y; Danckwardt, S. (2016). "Processing and transcriptome expansion at the mRNA 3′ end in health and disease: finding the right end". Eur J Physiol. 468 (6): 993–1012. doi:10.1007/s00424-016-1828-3. PMC 4893057. PMID 27220521.
| Genomics | |
|---|---|
| Bioinformatics | |
| Structural biology | |
| Research tools | |
| Organizations | |
| |