| Follicular lymphoma | |
|---|---|
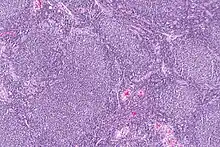 | |
| Micrograph of a follicular lymphoma, showing the characteristically abnormal lymphoid follicles that gave the condition its name. H&E stain. | |
| Specialty | Hematology and oncology |
Follicular lymphoma (FL) is a cancer that involves certain types of white blood cells known as lymphocytes. The cancer originates from the uncontrolled division of specific types of B-cells known as centrocytes and centroblasts. These cells normally occupy the follicles (nodular swirls of various types of lymphocytes) in the germinal centers of lymphoid tissues such as lymph nodes. The cancerous cells in FL typically form follicular or follicle-like structures (see adjacent Figure) in the tissues they invade. These structures are usually the dominant histological feature of this cancer.[1]
There are several synonymous and obsolete terms for FL such as CB/CC lymphoma (centroblastic and centrocytic lymphoma), nodular lymphoma,[2] Brill-Symmers Disease, and the subtype designation, follicular large-cell lymphoma.[3] In the US and Europe, this disease is the second most common form of non-Hodgkin's lymphomas, exceeded only by diffuse large B-cell lymphoma.[4] FL accounts for 10–20% of non-Hodgkin's lymphomas with ~15,000 new cases of it being newly diagnosed each year in the US and Europe.[5] Recent studies indicate that FL is similarly prevalent in Japan.[6]
FL is a broad and extremely complex clinical entity with a wide range of manifestations[7] which have not yet been fully systematized.[8] It is commonly preceded by a benign precancerous disorder in which abnormal centrocytes and/or centroblasts accumulate in lymphoid tissue. They may then circulate in the blood to cause an asymptomatic condition termed in situ lymphoid neoplasia of the follicular lymphoma type (i.e. ISFL). A small percentage of these cases progress to FL.[9] Most commonly, however, FL presents as a swelling of lymph nodes in the neck, armpits, and/or groin. Less often, it presents as a gastrointestinal tract cancer, a cancer in children involving lymphoid tissues of the head and neck area (e.g. tonsils),[10] or one or more masses in non-lymphoid tissues such as the testes.[11]
FL typically has a slow disease course which persists essentially unchanged for years.[7] However, each year 2–3%[12] of FL cases progress to a highly aggressive form often termed stage 3B FL, to an aggressive diffuse large B-cell lymphoma, or to another type of aggressive B-cell cancer. These transformed follicular lymphomas (t-FL) are essentially incurable.[5] However, recent advancements in the treatment of t-FL (e.g. the addition to standard chemotherapy of agents such as rituximab) have improved overall survival times. These newer regimens may also delay the transformation of FL to t-FL.[5] Additional advances in understanding FL may lead to further improvements in treating the disease.[12][13]
Pathophysiology
Genomic alterations
The serial progressions of in situ FL to FL and FL to t-FL appear to involve the accumulation of increasing numbers of genomic alterations (i.e. chromosome abnormalities and gene mutations) in the formative B-cell precursors to these disorders. At least some of these alterations appear to cause the over-expression or under-expression of the products of genes that regulate these cells' susceptibility to develop further genomic alterations, to survive, to proliferate, and/or to spread to other tissues. In consequence, multiple B-cell clones that exhibit increasing genomic alterations and malignant behaviors populate the disorder. No single genomic alteration seems responsible for the development of each of the spectrum of FL disorders. Rather, interactions between multiple genomic alterations appear to underlie this serial progression.[5][12]
In situ follicular lymphoma
In situ follicular lymphoma is an accumulation of monoclonal B cells (i.e. cells descendent from a single ancestral cell) in the germinal centers of lymphoid tissue. These cells commonly bear a pathological genomic abnormality, i.e. a translocation between position 32 on the long (i.e. "q") arm of chromosome 14 and position 21 on chromosome 18's q arm. This translocation juxtaposes the B-cell lymphoma 2 (BCL2) gene on chromosome 18 at position q21.33 near to the immunoglobulin heavy chain locus (IGH@) on chromosome 14 at position q32. In consequence, BCL2 overexpresses its product, BCL2 apoptosis regulator (i.e. Bcl2). Bcl2 functions to inhibit programmed cell death thereby prolonging cell survival.[14] The overexpression of Bcl2 in the B-cells of ISFL is thought to be a critical factor in their pathological accumulation and subsequent malignant progression.[9] Small numbers (e.g. 1 in 100,000) of circulating nucleated blood cells bearing this t(14:18)q32:q21) translocation are found in 50–67% of otherwise healthy individuals. The prevalence of this finding increases with age and years of tobacco smoking. Since most individuals with this translocation in their blood cells do not develop ISFL, the t(14:18)(q32:q21) translocation, while prolonging cell survival, must be just one step in the development of ISFN. This translocation is proposed to occur during the early development of immature bone marrow B-cells (i.e. pre-B-cells/pro-B-cells) after which these cells circulate freely and in rare cases accumulate and mature to centrocytes and/or centroblasts in the germinal centers of lymphoid follicles to form ISFL. The mechanism favoring this localization and further accumulation is unclear.[15]
Individuals with ISFL progress to FL at a rate of 2–3%/year for at least the first 10 years following diagnosis.[12] This progression likely involves the acquisition of genomic aberrations besides the t(14:18)q32:q21) translocation in the ISFL B-cells. Suspect mutations include those in the following genes: 1) EZH2 (encodes polycomb repressive complex 2 family protein which is involved in maintaining the transcriptional repressive state of various genes[16] and is found in up to 27% of FL cases);[9] 2) CREBBP (encodes CREB-binding protein which contributes to the activation of various genes[17]); 3) TNFSF14 (encodes tumor necrosis factor superfamily member 14, a member of the tumor necrosis factor superfamily which may function as a co-stimulatory factor for the activation of lymphoid cells[1][18]); and 4) KMT2D (encodes histone-lysine N-methyltransferase 2D, a histone methyltransferase which regulates the expression of various genes[19]).[20] ISFL may also acquire numerous copy-number variations (i.e. duplications and deletions of a portion of a chromosome along with any of the genes contained therein) that may contribute to FL. In all cases, the number of genetic abnormalities acquired in the B-cells of ISFL are much less than those in FL.[9]
Follicular lymphoma
The genomic alterations found in FL include 1) the t(14:18)(q32:q21.3) translocation (85–90% of cases); 2) 1p36 deletions (i.e. deletions in the q arm of chromosome 1 at position 36, [60–70% of cases]) that lead to lose of TNFAIP3 (encodes tumor necrosis factor, alpha-induced protein 3 which inhibits the activation of NF-κB, blocks cell death due to apoptosis, and regulates lymphocyte-based immune responses through its ubiquitin ligase activity[21]); 3) mutations in PRDM1 (encodes the PR domain zinc finger protein which promotes the maturation and proliferation of B-cells);[22] and 4) the same mutations seen in ISFL including KMT2D (85–90% of cases), CREEBP (40–65% of cases), BCL2 (40–65% of cases), and EZH2 (20–30% of cases) as well as other mutations such as those in the histone-modifying gene HIST1H1E (20–30% of cases), the RRAGC gene (~17% of cases) which regulates cell growth, survival, death, and proliferation,[23] and, in ≤15% of cases several other genes including MEF2B, STAT6, EP300, ARID1A, SLC22A2, CARD11, FOXO1, GNA12, B2M (i.e. the gene for beta-2 microglobulin), and SGK1. Except for the t(14:18)(q32:q21.3) translocation and EZH2 mutations which lead to gains in the expression and function, respectively, of their products, the genetic alterations generally lead to a loss in the production or function of the cited genes products. However, the exact roles, if any, of these genomic abnormalities in promoting the progression of ISFL to FL are unclear.[24]
Transformed follicular lymphoma
The transformation of FL to a more aggressive state or other type of aggressive lymphoma is associated with: 1) primarily gene-activating mutations in CREEBP, KMT2D, STAT6, CARD11 (encoding a guanylate kinase which interacts with BCL10 and activates NF-κB to regulate cell survival); 2) changes in the expression of diverse genes; 3) the overproduction of various cell-activating cytokines[25] and CD79B (encoding the Ig-beta protein component of the B-cell receptor[26]); 4) gene-inactivating mutations in TNFAIP3, CD58 (encoding the cell adhesion molecule, lymphocyte function-associated antigen 3, that is involved in activating T-cells[27]), CDKN2A (encoding p16INK4a and p14arf tumor suppressor proteins[28]) or CDKN2B (encoding cyclin dependent kinase inhibitor 2B multiple tumor suppressor 2[29]) (inactivation of either CDKN2 gene causes genome instability, i.e. increased frequency of other gene mutations), and TNFRSF4 (encoding one type of tumor necrosis factor receptor[30]); and 5) gene-activating or -inactivating mutations in, or other causes for the under- or over-expression of, c-MYC ((encoding the c-Myc proto-oncogene transcription factor that regulates the expression of diverse genes many of which promote cell proliferation[31]).[24]
Tumor environment
The non-neoplastic immune and stromal cells as well as the extracellular matrix in tissues may enable neoplastic follicular cells to survive, proliferate, and avoid surveillance by the immune system. For example, laboratory studies show that: 1) follicular dendritic cells, fibroblastic reticular cells, and T helper cells provide growth and survival signals to neoplastic follicular B-cells; 2) neoplastic follicular B-cells recruit regulatory T cells that act to suppress immune responses to them; 3) the cytotoxic T-cells which normally kill neoplastic cells become dysfunctional in the presence of neoplastic follicular cells that are embedded in this multicellular environment; and 4) bone marrow stromal cells directly support the growth of neoplastic follicular cells.[24] Reduced levels of immune-infiltration has been shown to be strongly associated with early progression of disease.[32]
Presentation and course
In situ follicular lymphoma
FL is commonly preceded by but uncommonly progresses to ISFL, an asymptomatic disorder that usually is discovered in tissues which are biopsied for other reasons. FL lymphoma may be diagnosed in the uncommon cases in which individuals with ISFL are found to have FL on follow-up examinations.[9] Similarly, individuals with >1 in 10,000 circulating lymphocytes containing the t(14:18)q32:q21) translocation are at increased but still small risk of developing FL and being diagnosed as having FL on follow up examinations.[10]
Follicular lymphoma
FL commonly presents as an otherwise asymptomatic enlargement of lymph nodes in the neck, armpit, groin,[13] femoral canal,[33] or other sites in individuals (median age 65) without a known history of ISFL or abnormal numbers of circulating t(14:18)q32:q21-conatianing lymphocytes.[13] These enlargements may have been present for months to years and during this time waxed and waned in size.[8] Less commonly, FL presents as extra-nodal masses in the skin, thyroid gland, salivary gland, breast, testicles.[11] spleen, liver,[33] and/or lung.[4] Regardless of the type of presentation, FL is usually (~80% of cases[8]) at an advanced stage at diagnosis as indicated by involvement of the bone marrow (50%[13] to 70%[8] of cases), multiple lymph nodes in different parts of the body,[9] and/or other tissues.[11] A minority (<33%)[8] of FL patients present with B symptoms, i.e. recurrent unexplained fevers, recurrent night sweats, and/or weight loss ≥10% in the past 6 months.[5] Generally, the disease has an indolent and prolonged course with a median life expectancy of 15–20 years: a large percentage of patients die from other causes than their FL disease.[5] However, each year, including the early years after diagnosis, some 2–3% of FL cases transform to t-FL;[12] Median survival has been ~4.5 years after the onset of this transformation.[5]
There are less common subtypes of FL that differ not only in their presentation but also in their histopathology, genetic abnormalities, and course. These subtypes, which are now (i.e. primary gastrointestinal tract FL) or may in the future (pediatric-type FL) be considered distinctive diseases, are:
Duodenal-type follicular lymphoma
Duodenal-type follicular lymphoma (DFL) was initially considered to be a type of Primary gastrointestinal tract (GI tract) follicular lymphoma (PGTFL), i.e. a follicular lymphoma in which GI tract lesions were prominent parts of the disease.[34] However, a subset of PGTFL cases had lesions that were localized to the duodenum and other parts of the small intestine usually without involving other parts of the GI tract or tissues outside of the GI tract. This contrasts with the other cases of PGTFL which were systemic diseases involving a wide range of GI tract and non-GI tract tissues. Consequently, the World Health Organization (2017) removed the localized disease from the primary gastrointestinal tract follicular lymphoma category, reclassified it as a distinct disease entity, and termed it duodenal-type follicular lymphoma.[6] DFL is most often an asymptomatic disease that is diagnosed on endoscopic examination of the GI tract conducted for other reasons. Less commonly, it presents with vague abdominal symptoms.[35][36] In one review of former studies, the lesions in 85% of primary duodenal follicular lymphoma were located not only in the duodenum but also other sites in the intestine (i.e. jejunum and/or ileum),[11] with rare cases having lesions in the rectum[37] or cecum[38] PDF is an indolent disease that may spontaneously remit and relapse but only rarely progresses to a more aggressive form. A watch-and-wait strategy has been a generally recommended method for the initial treatment of the disease.[39]
Primary gastrointestinal tract follicular lymphoma
PGTFL is a follicular lymphoma (which as currently defined excludes cases of duodenal-type follicular lymphoma) that has a prominent component of GI tract involvement. The disease may present with signs and symptoms typical of the common type of follicular lymphoma. For example, enlargement of lymph nodes in the neck, armpit, groin,[13] femoral canal, and/or other areas,[33] and/or signs and symptoms of GI tract disease[34] due to lesions in the stomach, small intestine, large intestine[11] or rectum may be seen.[37] These signs and symptoms may include abdominal pain, bowel obstruction,[11] persistent nausea and vomiting, hematochezia (i.e. passage of fresh blood usually on feces through the rectum), or melena (i.e. passage of tarry feces containing blood that has been digested in the stomach or upper intestine).[40] PGTFL is generally treated like cases of common follicular lymphoma: depending on the severity of the disease and its symptoms, patients are treated with watchful waiting, surgery, chemotherapy, radiation, immunotherapy plus radiotherapy, or combinations of these modalities.[40]
Predominantly diffuse follicular lymphoma with 1p36 deletion
Predominantly diffuse follicular lymphoma with 1p36 deletion is a rare subtype of FL[7] in which involved lymph nodes show infiltrations of centrocytes and centoblasts that generally do not form the nodular, swirling patterns characteristic of most types of FL.[1] In addition, these cells lack the t(14:18)(q32:q21.3) translocation commonly found in other FL types but, similar to many FL cases, have a deletion in the terminal part of the short (i.e. "p") arm of chromosome 1 that encodes the TNFRSF14 gene (see pathophysiology section).[13] Predominantly diffuse follicular lymphoma with 1p36 deletion usually presents with bulky enlargements of inguinal (i.e. groin) lymph nodes but may present with enlargements of the axillary (i.e. armpit) or cervical (i.e., neck) lymph nodes. In rare cases, there may be involvement of the bone marrow. In spite of the evidence of bulky and disseminated disease, predominantly diffuse follicular lymphoma with 1p36 deletion appears to be an indolent disorder that may require long-term observation rather than overtreatment.[7]
Pediatric-type follicular lymphoma
Pediatric-type follicular lymphoma (PTFL) was initially reported to occur in children ages 1–17 years old (median age ~13–14) but more recently has been reported to occur in adults.[41] The disorder was recently defined by the World Health Organization (2016) as a distinct entity that occurs mostly in males[7] and involves swollen lymph nodes in the head (including tonsils and adenoids), neck,[41] or, rarely, axillary, or inguinal areas, or non-lymphoid tissues.[42] Currently, however, patients who had exhibited or are exhibiting involvement of areas or tissues outside of the head, neck, armpit, or groin areas are now regarded as far more likely to have a newly and provisionally defined disease, large B-cell lymphoma with IRF4 rearrangement.[41]
The lesions in PTFL consists of infiltrates containing rapidly proliferating centrocytes and centroblasts that lack the t(14:18)(q32:q21.3) translocation but nonetheless often overexpress the BCL2 gene.[7] These cells may show a loss of heterozygosity at 1p36 (20-50% of cases) that results in decreased expression of the TNFRSF14 gene (see Pathophysiology section) as well as mutations in the IRF8 (10-50% of cases), which contributes to the development and function of B cells,[43][44] and the MAP2K1 gene (10–40% of cases), which regulates activation of the ERK cell signaling pathway.[45] More than 2 dozen other genes have been reported to be mutated in rare cases of PTFL but in general the genetic abnormalities found in this disorder are fewer and less complex than those in other types of FL.[42] PTFL has an indolent, relapsing and remitting course with a 5-year survival rate of >95%.[42] Patients diagnosed with PTFL have been treated with chemotherapy, surgery, and combinations of these treatments. In general, these patients did well (100% survival with <5% of cases relapsing regardless of treatment modality). More recently, 36 patients have been treated with surgical resection alone followed by observation; all these patients survived with only one having a relapse. Thus, PTFL appears to be a highly indolent type of FL in which multiple studies have reported overall and progression-free survival rates of 100% and >90%, respectively, for >2 years and an estimated probability of 5-year event-free survival rate of ~96%. The therapeutic regimens versus follow-up observations that best treat this disorder in children, adolescents, and adults (adults may require different treatments than children and adolescents) requires further study.[41]
Primary follicular lymphoma of the testis
Primary follicular lymphoma of the testis (PFLT), also termed testicular follicular lymphoma, was classified as a distinct form of FL by the World Health Organization in 2016.[33] It is an extremely rare disease that has been recognized as occurring primarily in children and adolescents[46] but also has been reported in 5 adults.[47] PFLT differs from cases of typical follicular lymphoma that involve the testis in that it more often occurs in children and adolescents; involves malignant B-cells that do have the t(14:18)q32:q21) translocation; and presents with disease that is strictly limited to the testis. While similar to pediatric-type follicular lymphoma in not involving cells that bear the t(14:18)q32:q21) translocation, PFLT differs from the former disease in that it is limited to the testis and involves malignant cells that do not express Bcl2.[48] PFTL is an extremely indolent disease which is manifested by lesions that exhibit a typical FL histology or, more commonly, a mixed FL-diffuse large cell lymphoma histology. It usually involves a 2–4 centimeter lesion in a single testicle. Patients have been treated with removal of the involved testes followed by various standard anti-lymphoma chemotherapy regimens to attain excellent results, i.e. 100% completed remissions with no recurrence of disease in 15 child and adolescent patients observed for 4–96 months. No cases of primary follicular lymphoma of the testis have been reported to progress to t-FL. Surgery followed by less strenuous or even no chemotherapy may prove to be the optimal treatment for this disease.[46]
Transformed follicular lymphoma
FL progresses at a rate of 2–3% per year for at least the first 10 years after diagnosis to a more aggressive form, principally diffuse large B-cell lymphoma (~93% of cases) or Burkitt-like lymphoma (~7% of cases) or in rare cases exhibit the histology resembling precursor B-cell lymphoblastic leukemia, plasmablastic lymphoma, the high grade subtype of B-cell lymphoma, Hodgkin lymphoma of the B-cell type, chronic lymphocytic leukemia/small cell lymphcytic lymphoma,[5] or histiocytic sarcoma.[1] t-FL is almost always diagnosed in patients being followed for FL. These FL patients present with the: fast growth of lymph nodes; formation of extra-nodal lesions in extra-nodal sites such as the central nervous system, liver or bone; the onset of B-symptoms (i.e. fever, night sweats, weight loss); development of hypercalcemia (i.e. high serum levels of calcium); and/or sudden rises in serum levels of the enzyme lactate dehydrogenase.[5] A minority of t-FL patients present without a history of FL. These patients generally present with advanced, bulky disease that may be accompanied by extra-nodal lesions and B-symptoms.[1] Typically, all the various forms of t-FL are aggressive, rapidly progressive diseases with overall media survival times in treated patients of ~4.5 years.[1][5] The transformation of FL to DLBCL is in over 70% of cases associated with the gain of MYC activity by genetic or non-genetic mechanisms.[49]
Diagnosis

The diagnosis of FL depends on examining involved tissues for histological, immunological, and chromosomal abnormalities that are indicative of the disease. FL usually involves enlarged lymph nodes populated by abnormal follicles (see adjacent picture) that when examined histologically contain a mixture of centrocytes or centroblast surrounded by non-malignant cells, mostly T-cells. The centrocytes, which typically outnumber centroblasts, are small to medium-sized B-cell lymphocytes that characteristically exhibit cleaved nuclei; the centropblasts are larger B-cell lymphocytes without cleaved nuclei.[11] Rare cases of FL may show lesions that contain tissue infiltrations dominated by B-cells with features of precursor (i.e. "blast") cells, monocytes, or malignant mantle cells such as those found in mantle cell lymphoma.[1] Immunochemical analyses reveal that these cells generally express B-cell surface markers including the CD10 (60% of cases), CD20, CD19, CD22, and CD79 but not CD5, CD11c, or CD23 cell surface proteins;[4] genomic analyses reveal that these cells contain t(14:18)(q32:q21.3) translocation (85–90% of cases), 1p36 deletions (60–70% of cases), and with far less frequency the other genomic abnormalities listed in the above sections on Pathophysiology and Presentation and course. None of these protein markers or genomic abnormalities are diagnostic for FL, e.g. the t(14:18)(q32:q21.3) translocation is found in 30% of diffuse large B-cell lymphoma and in a small number of reactive benign lymph nodes. Rather, the diagnosis is made by a combination of histological, immunological, and genomic abnormalities.[4] According to World Health Organization (WHO) criteria, follicular lymphoma can be classified morphologically by the relative amount of centroblasts. However, such classification is optional, due to poor reproducibility and little difference in prognosis and treatment, except that a lymphoma with almost only centroblasts may be diagnosed as a diffuse large B-cell lymphoma (DLBCL).[50] The optional classification of follicular lymphoma is as follows:[51]

- Centrocytes are small to medium size with angulated, elongated, cleaved, or twisted nuclei.
- Centroblasts are larger cells containing vesicular nuclei with one to three basophilic nucleoli apposing the nuclear membrane.
- Follicular dendritic cells have round nuclei, centrally located nucleoli, bland and dispersed chromatin, and flattening of adjacent nuclear membrane.
- Grade 1: follicles have <5 centroblasts per high-power field (hpf).
- Grade 2: follicles have 6 to 15 centroblasts per hpf.
- Grade 3: follicles have >15 centroblasts per hpf.
- Grade 3A: Grade 3 in which the follicles contain predominantly centrocytes.
- Grade 3B: Grade 3 in which the follicles consist almost entirely of centroblasts.
Grades 1 and 2 are regarded as low grade FL; Grade 3A is usually also regarded as low grade FL although some studies have regarded it as high grade FL; and Grade 3B is regarded as a highly aggressive FL in the t-FL category.[8]
In addition to grade 3B disease, histologic examinations may reveal other evidence of t-FL such as histologic findings consistent with FL and diffuse large cell lymphoma in the same tissue (referred to as composite lymphomas) or in separate tissues (referred to as (discordant lymphomas) or histologic findings similar to those found in Burkitt lymphoma, precursor B-cell lymphoblastic leukemia, plasmablastic lymphoma, the high grade subtype of B-cell lymphoma, Hodgkin lymphoma of the B-cell type, chronic lymphocytic leukemia/small cell lymphocytic lymphoma,[5] or histiocytic sarcoma.[1] Other findings indicating the presence of this transformation include rapid growth in size of lymph nodes, recently acquired or new B symptoms, recent development of FL lesions in non-nodal tissue, rapid rises in serum lactate dehydrogenase levels, and the presence of high levels of serum calcium.[12]
Differential diagnosis
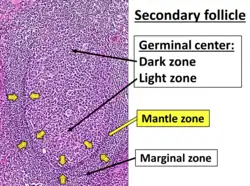
FL may be confused with marginal zone B-cell lymphoma, mantle cell lymphoma, and the small lymphocytic lymphoma variant of chronic lymphocytic leukemia. The malignant cells in marginal zone B-cell lymphoma may form follicular structures but commonly proliferate in the marginal zone rather than germinal center of lymphoid tissues. These malignant cells often show features of monocytes or plasma cells. Mantle cell lymphomas show monotonous, medium-sized lymphocytes, monocytes, and atrophied germinal centers; unlike FL, the malignant lymphocytes in this disease are positive for Cyclin D1 by immunohistochemistry staining. Small lymphocytic lymphomas are composed of nodular structures with small- to medium-sized malignant cells surrounding immature lymphocytes and immunoblasts. The malignant cells in this disease, unlike FL, stain positive for CD5 and CD23.[11]
Treatment and prognosis
FL is typically a slowly growing lymphoma with an overall median life expectancy for treated patients of 10–15 years[34] with many cases of it waxing and waning in the size of their lesions and rare cases of it remitting spontaneously.[4] These considerations favor the use of observation over intervention in patients whose particular form of FL has a favorable prognosis or who are intolerant to aggressive treatments.[4] However, most cases of FL have a less favorable prognosis at some stage of their disease and will therefore require intervention. There is little consensus regarding the guidelines to be used to define the prognosis and treatment for FL at its presentation or during its course.. Currently used indicators for this include the disease's: 1) histology; 2) subtype; 3) predicted indolence and potential for transformation; and 4) extent of disease as measured by clinical examinations, bone marrow biopsy to determine bone marrow involvement, and PET/CT imaging of the chest, abdomen, pelvis, and any areas outside of these regions if physical examination suggests involvement.[52] Some suggested guidelines using these parameters to indicate the prognosis and need for treatment in FL include:[8]
- The WHO criteria using histological grade (see previous section): Patients with Grades 1, 2, and 3A disease are predicted to have the same low risk prognosis that is seen in cases of typical FL while patients with grade 3B disease are predicted to have the high risk prognosis typical of t-FL.
- The Follicular Lymphoma International Prognostic Index (FLIPI): FLIPI uses the following criteria: age ≥60 years; Ann Arbor disease stage III (i.e. lesions located both above and below the thoracic diaphragm) or IV (i.e. disseminated lesions involving one or more non-lymphatic organs); blood hemoglobin <12 gram/deciliter; serum lactose dehydrogenase level above normal; and involvement of >4 lymph nodes. Patients positive for 0–1, 2, or ≥3 of these factors are classified as in low, intermediate, and high risk group, respectively, and after treatment with regimens that include rituximab have 2 year predicted progression free survivals of 84, 72, and 65%, respectively, and overall survivals of 98, 94, and 87%, respectively.[4]
- The FLIP2 index. This modification of FLIP1 uses age ≥60; blood hemoglobin <12 gram/deciliter; serum lactose dehydrogenase level above normal; serum beta-2 microglobulin level above normal; ≥1 lymph node with a diameter >6 centimeters; and bone marrow involvement. The predicted percentage of therapy-treated patients with progression free survival at 5 years for individuals positive for 0, 1–2, and ≥3 of these factors are 80, 51, and 19%, respectively.[8]
- CT/PET imaging: This method measures total body tumor volume as detected by tissue uptake of radioactive fludeoxyglucose (F18). Progression free and overall survival at 5 years for patients with estimated tumor volumes above versus below 510 cubic centimeters are reported to be 32.7 and 84.8% versus 65.1 and 94.7%, respectively.[8]
- Lugano staging: this method classifies Stage I disease as involving a single lymphatic region or extra-lymphatic site; Stage II disease as involving ≥2 lymphatic sites or 1 lymphatic site plus 1 extralympatic site with all lesions being on the same side of the diaphragm; Stage III disease as involving ≥2 lymphatic regions that are on opposite sides of the diaphragm; and Stage IV disease as disseminated lesions that are found to be in ≥1 non-lymphatic organs.[4]
- Response-based prognosis: FL patients whose disease progresses within 24 months of initiating treatment with chemotherapy and immunotherapy versus patients whose disease does not progress within 24 months are predicted to have 5 year survival rates of 50–74% versus ~90%, respectively.[8]
The prognosis and treatment for the specific presentations of typical FL cases (see above sections for the prognoses and treatment recommendations for primary gastrointestinal tract FL, predominantly diffuse FL with 1p36 deletion, pediatric-type FL, and primary FL of the testis) that are in common use are as follows:
In situ follicular lymphoma
ISFL is a benign condition that may be reevaluated periodically to detect the rare cases of it which progress to FL; otherwise ISFL is not treated.[9]
Localized follicular lymphoma
In 10–20% of cases, FL appears limited to single radiation field, does not involve the bone marrow, and is therefore regarded as localized early-stage FL. In these cases, which are sometimes classified as Ann Arbor stage I (i.e. disease limited to a single restricted region) or stage II (i.e. disease restricted to two sites that are on the same side of the diaphragm),[4] radiation therapy achieves 10 year overall survival rates of 60–80% and median overall survival times of 19 years.[8] It seems likely that many of the relapses in these cases are due to undetected disease outside of the radiation field at the time of radiation treatment. The use of PET/CT imaging is strongly recommended to insure that the FL is localized. In any case, the excellent results achieved with radiation therapy strongly support its use in localized disease. The use of an immunotherapeutic agent such as Rituximab alone or in combination with a chemotherapeutic regimen such as CVP (i.e. cyclophosphamide, vincristine, prednisone and rituximab) in cases of localized, early-stage disease may be appropriate choices for some of these early-stage patients.[4] However, the latter approach is recommended for cases of localized disease in which the disease extends beyond a single field: 56% of patients treated in this manner had progression-free survival at 10 years while patients treated with other regimens had progression free survivals of 41%. Nonetheless, overall survival did not differ between the two groups.[13]
Asymptomatic follicular lymphoma
Patients with asymptomatic but not localized low grade FL,[8][53][54] gastrointestinal tract FL,[34] and pediatric-type follicular lymphoma[41] have been served by careful follow-up without therapeutic intervention. Even high grade, aggressive, relapsed, or transformed FL may also be served with observation in patients who are asymptomatic. Findings in asymptomatic patients who have been recommended as triggers for starting treatment include one or more of the following: tumor size ≥7 cm in diameter; involvement of ≥3 nodes in 3 distinct areas, each of which is ≥3 cm in diameter; organ compression; presence of ascites or pleural effusion (i.e. build-up of fluid in the abdominal or pleural cavities); poor performance status due to the disease; elevated levels of serum lactose dehydrogenase or beta-2 microglobulin;[4] presence of localized bone lesions; kidney involvement; reduced levels of circulating blood platelets or any of the various types of white blood cells; onset of significant pruritus (i.e. itching sensation) or other B symptoms; and enlargement (i.e. ≥50% increase in size over a period of at least 6 months) of lymph nodes, spleen, or other follicular lymphoma-infiltrated organs or tissues.[33]
Symptomatic follicular lymphoma
Symptomatic FL requires treatments directed at relieving symptoms by reducing the load of tumor cells. Various chemotherapeutic regimens have been used for this including combinations of alkylating antineoplastic agents, nucleoside analogues, and/or anthracyclines. Two commonly used chemotherapeutic regimens are CVP (see Localized FL section) and CHOP (i.e. CVP plus the anthracycline adriamycin). Newer agents used to treat FL include monoclonal antibodies such as rituximab, obinutuzumab, galiximab, inotuzumab ozogamicin, or epratuzumab and immunomodulators such as lenalidomide and interferon. The latter medications have been used in combination or alone to treat symptomatic FL.[13] Most such regimens add rituximab (a monoclonal antibody which binds and thereby kills the CD20 cell surface protein on B cells) with CVP or CHOP regimens (termed R-CVP and R-CHOP regimens).
The R-CHOP regimen appears superior to the R-CVP regimen with, for example, one study finding 8-year progression-free survival rates of 57% versus 46% for the two respective regimens.[33] More recently, FL patients have been treated with other regimens including: 1) rituximab combined with the chemotherapeutic alkylating agent bendamustine; 2) rituximab combined with the chemotherapeutic agent fludarabine and the inhibitor of Type II topoisomerase, mitoxantrone;[33] and 3) rituximab combined with another immunotherapeutic agent such as galiximab, epratuzumab (monoclonal antibodies directed respectively against the CD80 or CD22 cell surface proteins on immune cells including B cells), or the immunomodulating medication, lenalidomide.[13] While it is too soon to judge the long-term results of the latter regimens, the regimens have shown similar results when analyzed based on poor treatment responses (~10–20% poor responses). Bendamustine with rituximab may be preferable to R-CHOP or R-CVP for treating low-grade (i.e. Grades 1, 2, and possibly 3A) FL; R-CHOP may be preferred in FL that has high-risk characteristics (e.g. high levels of Beta-2 macroglobulin or bone marrow involvement). The combination of lenalidomide with rituximab has shown good potential in treating indolent cases of FL.[13]
Studies indicate that maintenance therapy with rituximab following successful induction therapy prolongs progression-free survival; for example one study found progression-free survival after 6 years of treatment was 59.2% in patients treated with rituximab maintenance and 42.7% without this maintenance; however, overall survival at 6 years was similar in the two groups, 87.4% and 88.7%, respectively. Another study found that prolonged maintenance with rituximab did not have any benefits over an eight-month maintenance period.[13] Finally, surgery[55][56] and radiation[4][13][33] are additional therapies that can be used to relieve symptoms caused by bulky t-FL disease or to treat lesions in patients who cannot withstand other types of treatment.
Transformed follicular lymphoma
Early studies on treating t-FL with various purely chemotherapy regimens gave poor results with median overall survival times of 1–2 years. However, the addition of rituximab to the regimens such as CVP and CHOP as part of induction and maintenance therapies (i.e. R-CVP and R-CHOP) greatly improved overall 5 year survival to rates of 73%. The R-CHOP regimen is a good option for treating such cases.[5] However, these regimens need not be started in people with FL who are asymptomatic and have low tumor burdens: the outcomes in such patients show no difference between early versus delayed treatment. Some recent studies found that the use of rituximab in combination with bendamustine (i.e. the RB regimen) provided better results than R-CHOP: progression-free survival times in one study were 69.5 months for RB and 31.2 months for R-CHOP. Similar results were obtained when RB was compared to R-CVP. These studies also found no overall survival time benefit between the RB and R-CHOP regimens. Other recently examined regimens include 1) the use of obinutuzumab instead of rituximab in the R-CHOP and R-CVP regiments to attain progression-free survival rates at 3 years of 80% for the obinutuzumab-chemotherapy regimen versus 73% for the rituximab-chemotherapy regimen and 2) the combination of rituximab with lenalidomide (no chemotherapy agent) versus various chemotherapy plus immunotherapy (principally rituximab) to achieve similar complete remission and 3 year progression-free survival rates but with rituximab plus lenalidomide causing less toxicity (i.e. severe neutropenia). Many of these studies did use rituximab maintenance therapy after induction therapy.[4]
Prevention
Several studies, while not conclusive, suggest that the early treatment of low risk FL reduces the incidence of the disease progressing to t-FL. The treatments used in these studies include chemotherapy, radiation therapy, and immunotherapy combinations plus rituximab maintenance therapy.[12]
Relapsed follicular lymphoma
Patients who relapse after initial therapy for FL may be followed closely without therapy if asymptomatic. When treatment is required, patients may be treated with the initial treatment regimen when such treatment led to a remission that lasted for at least one year; otherwise an alternative regimen is used.[13] The regimens commonly used in relapsed lymphoma include R-CHOP, R-CVP, RFM (i.e. rituximab, fludarabine, and mitoxantrone), and RB (Bendamustine plus rituximab).[4] Patients who have early treatment failure (e.g. within 1–2 years of initial treatment) or multiple relapses have also been treated with either autologous (i.e. stem cells taken from patient) or allogeneic (i.e. stem cells taken from a donor) stem cell bone marrow transplantation. While studies are inconclusive, autologous stem cell bone marrow transplantation appears to prolong survival in early treatment failure patients who are healthy enough to withstand this therapy. Unfit patients may benefit from initial treatment with obinutuzumab plus bendamustine followed by maintenance treatment with obinutuzumab (if they have not been treated previously with obinutuzumab).[13]
Other mostly experimental treatments currently under study in patients with multiple treatment failures include: 1) Phosphoinositide 3-kinase inhibitors such as copanlisib, duvelisib, and idelalisib which block the phosphoinositide 3-kinase signaling pathway that promotes the survival, proliferation, and other potentially malignant behaviors of cells; 2) infusion of tisagenlecleucel chimeric antigen receptor T cells (i.e. CAR T cells) (i.e. T cells that have been isolated from patients, engineered to express a receptor for the CD19 protein on, and thereby kill, T cells, and then infused back into the donor patient);[52] 3) Bruon's tyrosine kinase inhibitor, ibrutinib, to block the B-cell maturating actions of this kianase; 4) BCL inhibitor venetoclax to block Bcl2's action in promoting B-cell survival and proliferation; 5) histone deacetylase inhibitors abexinostat and tazemetostat to modify the expression of various genes; and 6) Checkpoint inhibitors nivolumab, pidilizumab, and pembrolizumab to promote the immune system's ability to suppress cancer cell growth.[4] In preliminary studies on FL patients who were known or thought to be refractor to more conventional therapies these drugs, when combined with more conventional drugs, particularly rituximab, produced promising results. Phosphoionsitide 3-kinase inhibitors produced overall response rates of 10–12.5 months in 42–59%; tisagenlecleuce cells produced an overall progression-free response rate of 70% after a follow-up of 28 months;[52] phosphoinositide 3-kinase inhibitors produced overall response rates of ~40% and complete response rates of 1–20%; Bruton's tyrosine kinase inhibitor produced overall and complete response rates of 38% and 18%, respectively; the Bcl inhibitor produce overall and complete response rates of 33% and 14%, respectively; histone deacetylase inhibitors produce overall response rates of 35–71%; and checkpoint inhibitors produce overall response rates of 40–80% and complete response rates of 10–60%.[4]
See also
References
- 1 2 3 4 5 6 7 8 Xerri L, Dirnhofer S, Quintanilla-Martinez L, Sander B, Chan JK, Campo E, et al. (February 2016). "The heterogeneity of follicular lymphomas: from early development to transformation". Virchows Archiv. 468 (2): 127–39. doi:10.1007/s00428-015-1864-y. PMID 26481245. S2CID 2978889.
- ↑ "follicular lymphoma""at Dorland's Medical Dictionary
- ↑ Large-Cell+Lymphoma,+Follicular at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Dada R (June 2019). "Diagnosis and management of follicular lymphoma: A comprehensive review". European Journal of Haematology. 103 (3): 152–163. doi:10.1111/ejh.13271. PMID 31270855.
- 1 2 3 4 5 6 7 8 9 10 11 12 Fischer T, Zing NP, Chiattone CS, Federico M, Luminari S (January 2018). "Transformed follicular lymphoma". Annals of Hematology. 97 (1): 17–29. doi:10.1007/s00277-017-3151-2. hdl:11380/1152780. PMID 29043381. S2CID 11524500.
- 1 2 Yoshino T, Takata K, Tanaka T, Sato Y, Tari A, Okada H (December 2018). "Recent progress in follicular lymphoma in Japan and characteristics of the duodenal type". Pathology International. 68 (12): 665–676. doi:10.1111/pin.12733. PMID 30456840. S2CID 53871784.
- 1 2 3 4 5 6 Lynch RC, Gratzinger D, Advani RH (July 2017). "Clinical Impact of the 2016 Update to the WHO Lymphoma Classification". Current Treatment Options in Oncology. 18 (7): 45. doi:10.1007/s11864-017-0483-z. PMID 28670664. S2CID 4415738.
- 1 2 3 4 5 6 7 8 9 10 11 12 Boughan KM, Caimi PF (May 2019). "Follicular Lymphoma: Diagnostic and Prognostic Considerations in Initial Treatment Approach". Current Oncology Reports. 21 (7): 63. doi:10.1007/s11912-019-0808-0. PMID 31119485. S2CID 162181232.
- 1 2 3 4 5 6 7 Oishi N, Montes-Moreno S, Feldman AL (January 2018). "In situ neoplasia in lymph node pathology". Seminars in Diagnostic Pathology. 35 (1): 76–83. doi:10.1053/j.semdp.2017.11.001. PMID 29129357.
- 1 2 Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES (May 2016). "The 2016 revision of the World Health Organization classification of lymphoid neoplasms". Blood. 127 (20): 2375–90. doi:10.1182/blood-2016-01-643569. PMC 4874220. PMID 26980727.
- 1 2 3 4 5 6 7 8 Takata K, Miyata-Takata T, Sato Y, Yoshino T (2014). "Pathology of follicular lymphoma". Journal of Clinical and Experimental Hematopathology. 54 (1): 3–9. doi:10.3960/jslrt.54.3. PMID 24942941.
- 1 2 3 4 5 6 7 Link BK (March 2018). "Transformation of follicular lymphoma – Why does it happen and can it be prevented?". Best Practice & Research. Clinical Haematology. 31 (1): 49–56. doi:10.1016/j.beha.2017.10.005. PMID 29452666.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Sorigue M, Sancho JM (February 2018). "Current prognostic and predictive factors in follicular lymphoma". Annals of Hematology. 97 (2): 209–227. doi:10.1007/s00277-017-3154-z. PMID 29032510. S2CID 27602442.
- ↑ EntrezGene 596
- ↑ Karube K, Scarfò L, Campo E, Ghia P (February 2014). "Monoclonal B cell lymphocytosis and "in situ" lymphoma". Seminars in Cancer Biology. 24: 3–14. doi:10.1016/j.semcancer.2013.08.003. PMID 23999128.
- ↑ EntrezGene 2146
- ↑ EntrezGene 1387
- ↑ EntrezGene 8740
- ↑ EntrezGene 8085
- ↑ Carbone A, Gloghini A (March 2014). "Emerging issues after the recognition of in situ follicular lymphoma". Leukemia & Lymphoma. 55 (3): 482–90. doi:10.3109/10428194.2013.807926. PMID 23713483. S2CID 39451928.
- ↑ EntrezGene 7128
- ↑ EntrezGene 639
- ↑ EntrezGene 64121
- 1 2 3 Gascoyne RD, Nadel B, Pasqualucci L, Fitzgibbon J, Payton JE, Melnick A, et al. (December 2017). "Follicular lymphoma: State-of-the-art ICML workshop in Lugano 2015". Hematological Oncology. 35 (4): 397–407. doi:10.1002/hon.2411. hdl:11343/292747. PMID 28378425. S2CID 23980925.
- ↑ EntrezGene 84433
- ↑ EntrezGene 974
- ↑ EntrezGene 965
- ↑ EntrezGene 1029
- ↑ EntrezGene 1030
- ↑ EntrezGene 8764
- ↑ EntrezGene 4609
- ↑ Tobin JW, Keane C, Gunawardana J, Mollee P, Birch S, Hoang T, Lee J, Li L, Huang L, Murigneux V, Fink JL, Matigian N, Vari F, Francis S, Kridel R, Weigert O, Haebe S, Jurinovic V, Klapper W, Steidl C, Sehn LH, Law S, Wykes MN, and Gandhi MK (December 2019). "Progression of Disease Within 24 Months in Follicular Lymphoma Is Associated With Reduced Intratumoral Immune Infiltration". J Clin Oncol. 37 (34): 3300–3309. doi:10.1200/JCO.18.02365. PMC 6881104. PMID 31461379.
- 1 2 3 4 5 6 7 8 Bargetzi M, Baumann R, Cogliatti S, Dietrich PY, Duchosal M, Goede J, Hitz F, Konermann C, Lohri A, Mey U, Novak U, Papachristofilou A, Stenner F, Taverna C, Zander T, Renner C (2018). "Diagnosis and treatment of follicular lymphoma: an update". Swiss Medical Weekly. 148: w14635. doi:10.4414/smw.2018.14635. PMID 30044476.
- 1 2 3 4 Takata K, Miyata-Takata T, Sato Y, Iwamuro M, Okada H, Tari A, Yoshino T (January 2018). "Gastrointestinal follicular lymphoma: Current knowledge and future challenges". Pathology International. 68 (1): 1–6. doi:10.1111/pin.12621. PMID 29292593. S2CID 206275496.
- ↑ Foukas PG, de Leval L (January 2015). "Recent advances in intestinal lymphomas". Histopathology. 66 (1): 112–36. doi:10.1111/his.12596. PMID 25639480. S2CID 20669863.
- ↑ Lightner AL, Shannon E, Gibbons MM, Russell MM (April 2016). "Primary Gastrointestinal Non-Hodgkin's Lymphoma of the Small and Large Intestines: a Systematic Review". Journal of Gastrointestinal Surgery. 20 (4): 827–39. doi:10.1007/s11605-015-3052-4. PMID 26676930. S2CID 21756793.
- 1 2 Pyeon SI, Song GA, Baek DH, Kim GH, Lee BE, Lee SJ, Yoon JB, Han SY, Park DY (February 2017). "Primary Follicular Lymphoma in the Rectum Incidentally Found on Screening Colonoscopy". The Korean Journal of Gastroenterology = Taehan Sohwagi Hakhoe Chi. 69 (2): 139–142. doi:10.4166/kjg.2017.69.2.139. PMID 28239083.
- ↑ Marks E, Shi Y (April 2018). "Duodenal-Type Follicular Lymphoma: A Clinicopathologic Review". Archives of Pathology & Laboratory Medicine. 142 (4): 542–547. doi:10.5858/arpa.2016-0519-RS. PMID 29565210.
- ↑ Weindorf SC, Smith LB, Owens SR (November 2018). "Update on Gastrointestinal Lymphomas". Archives of Pathology & Laboratory Medicine. 142 (11): 1347–1351. doi:10.5858/arpa.2018-0275-RA. PMID 30407861.
- 1 2 Moy BT, Wilmot J, Ballesteros E, Forouhar F, Vaziri H (September 2016). "Primary Follicular Lymphoma of the Gastrointestinal Tract: Case Report and Review". Journal of Gastrointestinal Cancer. 47 (3): 255–63. doi:10.1007/s12029-016-9847-z. PMID 27277664. S2CID 1014977.
- 1 2 3 4 5 Woessmann W, Quintanilla-Martinez L (June 2019). "Rare mature B-cell lymphomas in children and adolescents". Hematological Oncology. 37 (Suppl 1): 53–61. doi:10.1002/hon.2585. PMID 31187530.
- 1 2 3 Koo M, Ohgami RS (May 2017). "Pediatric-type Follicular Lymphoma and Pediatric Nodal Marginal Zone Lymphoma: Recent Clinical, Morphologic, Immunophenotypic, and Genetic Insights". Advances in Anatomic Pathology. 24 (3): 128–135. doi:10.1097/PAP.0000000000000144. PMID 28277421.
- ↑ Shukla V, Lu R (August 2014). "IRF4 and IRF8: Governing the virtues of B Lymphocytes". Frontiers in Biology. 9 (4): 269–282. doi:10.1007/s11515-014-1318-y. PMC 4261187. PMID 25506356.
- ↑ "IRF8 interferon regulatory factor 8 [Homo sapiens (human)] – Gene – NCBI".
- ↑ "MAP2K1 mitogen-activated protein kinase kinase 1 [Homo sapiens (human)] – Gene – NCBI".
- 1 2 Lones MA, Raphael M, McCarthy K, Wotherspoon A, Terrier-Lacombe MJ, Ramsay AD, Maclennan K, Cairo MS, Gerrard M, Michon J, Patte C, Pinkerton R, Sender L, Auperin A, Sposto R, Weston C, Heerema NA, Sanger WG, von Allmen D, Perkins SL (January 2012). "Primary follicular lymphoma of the testis in children and adolescents". Journal of Pediatric Hematology/Oncology. 34 (1): 68–71. doi:10.1097/MPH.0b013e31820e4636. PMC 3251817. PMID 22215099.
- ↑ Xu H, Yao F (March 2019). "Primary testicular lymphoma: A SEER analysis of 1,169 cases". Oncology Letters. 17 (3): 3113–3124. doi:10.3892/ol.2019.9953. PMC 6396186. PMID 30867741.
- ↑ Cheah CY, Wirth A, Seymour JF (January 2014). "Primary testicular lymphoma". Blood. 123 (4): 486–93. doi:10.1182/blood-2013-10-530659. PMID 24282217.
- ↑ Lossos, I. S.; Gascoyne, R. D. (2011). "Transformation of follicular lymphoma". Best Practice & Research. Clinical Haematology. 24 (2): 147–63. doi:10.1016/j.beha.2011.02.006. PMC 3112479. PMID 21658615.
- ↑ Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E; et al. (2022). "The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms". Leukemia. 36 (7): 1720–1748. doi:10.1038/s41375-022-01620-2. PMC 9214472. PMID 35732829.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Weissmann D. "Follicular Lymphomas". University of Medicine and Dentistry of New Jersey. Archived from the original on 2016-03-04. Retrieved 2008-07-26.
- 1 2 3 Sorigue M, Sancho JM (May 2019). "Recent landmark studies in follicular lymphoma". Blood Reviews. 35: 68–80. doi:10.1016/j.blre.2019.03.006. PMID 30928169. S2CID 89617933.
- ↑ Lister A. "Follicular Lymphoma: Perspective, Treatment Options, and Strategy". MedScape.
- ↑ Solal-Céligny P, Bellei M, Marcheselli L, Pesce EA, Pileri S, McLaughlin P, Luminari S, Pro B, Montoto S, Ferreri AJ, Deconinck E, Milpied N, Gordon LI, Federico M (November 2012). "Watchful waiting in low-tumor burden follicular lymphoma in the rituximab era: results of an F2-study database". Journal of Clinical Oncology. 30 (31): 3848–53. doi:10.1200/JCO.2010.33.4474. PMID 23008294.
- ↑ Ganapathi KA, Pittaluga S, Odejide OO, Freedman AS, Jaffe ES (September 2014). "Early lymphoid lesions: conceptual, diagnostic and clinical challenges". Haematologica. 99 (9): 1421–32. doi:10.3324/haematol.2014.107938. PMC 4562530. PMID 25176983.
- ↑ Pavanello F, Steffanoni S, Ghielmini M, Zucca E (2016). "Systemic Front Line Therapy of Follicular Lymphoma: When, to Whom and How". Mediterranean Journal of Hematology and Infectious Diseases. 8 (1): e2016062. doi:10.4084/MJHID.2016.062. PMC 5111519. PMID 27872742.
External links
- Follicular large cell lymphoma entry in the public domain NCI Dictionary of Cancer Terms