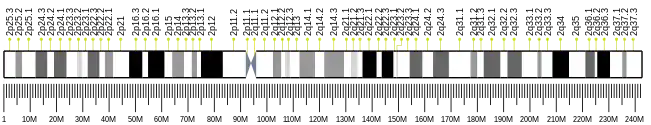



The PAX3 (paired box gene 3) gene encodes a member of the paired box or PAX family of transcription factors.[5] The PAX family consists of nine human (PAX1-PAX9) and nine mouse (Pax1-Pax9) members arranged into four subfamilies. Human PAX3 and mouse Pax3 are present in a subfamily along with the highly homologous human PAX7 and mouse Pax7 genes. The human PAX3 gene is located in the 2q36.1 chromosomal region, and contains 10 exons within a 100 kb region.
Transcript splicing
Alternative splicing and processing generates multiple PAX3 isoforms that have been detected at the mRNA level.[6] PAX3e is the longest isoform and consists of 10 exons that encode a 505 amino acid protein. In other mammalian species, including mouse, the longest mRNAs correspond to the human PAX3c and PAX3d isoforms, which consist of the first 8 or 9 exons of the PAX3 gene, respectively. Shorter PAX3 isoforms include mRNAs that skip exon 8 (PAX3g and PAX3h) and mRNAs containing 4 or 5 exons (PAX3a and PAX3b). In limited studies comparing isoform expression, PAX3d is expressed at the highest levels. From a functional standpoint, PAX3c, PAX3d, and PAX3h stimulate activities such as cell growth whereas PAX3e and PAX3g inhibit these activities, and PAX3a and PAX3b show no activity or inhibit these endpoints.
A common alternative splice affecting the PAX3 mRNA involves the sequence CAG at the 5’ end of exon 3.[7] This splice either includes or excludes these three bases, thus resulting in the presence or absence of a glutamine residue in the paired box motif. Limited sequencing studies of full-length human cDNAs identified this splicing event as a variant of the PAX3d isoform, and this spliced isoform has been separately termed the PAX3i isoform. The Q+ and Q- isoforms of PAX3 are generally co-expressed in cells. At the functional level, the Q+ isoform shows similar or less DNA binding and transcriptional activation than the Q- isoform.
Protein structure and function

PAX3 encodes a transcription factor with an N-terminal DNA binding domain consisting of a paired box (PD) encoded by exons 2, 3, and 4, and an octapeptide and complete homeodomain (HD) encoded by exons 5 and 6.[8] In addition, the PAX3 protein has a C-terminal transcriptional activation domain encoded by exons 7 and 8. The highly conserved PD consists of a 128 amino acid region that binds to DNA sequences related to the TCACGC/G motif.[9] The HD motif usually consists of 60 amino acids and binds to sequences containing a TAAT core motif.[10] The combination of these two DNA binding domains enable the PAX3 protein to recognize longer sequences containing PD and HD binding sites.[11] In the C-terminus of PAX3, there is a proline, serine and threonine (PST)-rich region measuring 78 amino acids that functions to stimulate transcriptional activity.[12] There are also transcriptional repression domains in the HD and N-terminal region (including the first half of the PD) that repress the C-terminal transcriptional activation domain.[13]
PAX3 functions as a transcriptional activator for most target genes, but also may repress a smaller subset of target genes.[14] These expression changes are effected through binding of PAX3 to specific recognition sites, which are situated in various genomic locations.[15] Some binding sites are located in or near target genes, such as the 5’ promoter, first intron and 3’ untranslated region. A substantial number of PAX3 binding sites are located at larger distances upstream and downstream of target genes. Among the PAX3 target genes, there is one group associated with muscle development and a second group associated with neural and melanocyte development. The proteins encoded by these target genes regulate various functional activities in these lineages, including differentiation, proliferation, migration, adhesion, and apoptosis.
PAX3 interacts with other nuclear proteins, which modulate PAX3 transcriptional activity. Dimerization of PAX3 with another PAX3 molecule or a PAX7 molecule enables binding to a palindromic HD binding site (TAATCAATTA).[16] Interaction of PAX3 with other transcription factors (such as SOX10) or chromatin factors (such as PAX3/7BP) enables synergistic activation of PAX3 target genes.[17][18] In contrast, binding of PAX3 to co-repressors, such as calmyrin, inhibits activation of PAX3 target genes.[19] These co-repressors may function by altering chromatin structure at target genes, inhibiting PAX3 recognition of its DNA binding site or directly altering PAX3 transcriptional activity.
Finally, PAX3 protein expression and function can be modulated by post-translational modifications. PAX3 can be phosphorylated at serines 201, 205 and 209 by kinases such as GSK3b, which in some settings will increase PAX3 protein stability.[20][21] In addition, PAX3 can also undergo ubiquitination and acetylation at lysines 437 and 475, which regulates protein stability and function.[22][23]
Table 1. Representative PAX3 transcriptional target genes.
| Protein category | Name | Phenotypic Activity |
| Cell adhesion molecule | NRCAM | Intercellular adhesion |
| Chemokine receptor | CXCR4 | Motility |
| Receptor tyrosine kinase | FGFR4 | Proliferation, differentiation, migration |
| MET | Proliferation, migration, survival | |
| RET | Proliferation, migration, differentiation | |
| Transcription factor | MITF | Differentiation, proliferation, survival |
| MYF5 | Differentiation | |
| MYOD1 | Differentiation |
Expression during development
During development, one of the major lineages expressing Pax3 is the skeletal muscle lineage.[24] Pax3 expression is first seen in the pre-somitic paraxial mesoderm, and then ultimately becomes restricted to the dermomyotome, which forms from the dorsal region of the somites. To form skeletal muscle in central body segments, PAX3-expressing cells detach from the dermomyotome and then Pax3 expression is turned off as Myf5 and MyoD1 expression is activated. To form other skeletal muscles, Pax3-expressing cells detach from the dermomyotome and migrate to more distant sites, such as the limbs and diaphragm. A subset of these Pax3-expressing dermomyotome-derived cells also serves as an ongoing progenitor pool for skeletal muscle growth during fetal development. During later developmental stages, myogenic precursors expressing Pax3 and/or Pax7 form satellite cells within the skeletal muscle, which contribute to postnatal muscle growth and muscle regeneration. These adult satellite cells remain quiescent until injury occurs, and then are stimulated to divide and regenerate the injured muscle.
Pax3 is also involved in the development of the nervous system.[25] Expression of Pax3 is first detected in the dorsal region of the neural groove and, as this neural groove deepens to form the neural tube, Pax3 is expressed in the dorsal portion of the neural tube. As the neural tube enlarges, Pax3 expression is localized to proliferative cells in the inner ventricular zone and then this expression is turned off as these cells migrate to more superficial regions. Pax3 is expressed along the length of the neural tube and throughout much of the developing brain, and this expression is subsequently turned off during later developmental stages in a rostral to caudal direction.
During early development, Pax3 expression also occurs at the lateral and posterior margins of the neural plate, which is the region from which the neural crest arises.[26] Pax3 is later expressed by various cell types and structures arising from the neural crest, such as melanoblasts, Schwann cell precursors, and dorsal root ganglia. In addition, Pax3-expressing cells derived from the neural crest contribute to the formation of other structures, such as the inner ear, mandible and maxilla.[27]
Pax3 controls the location of the nasion (a facial feature between the eyes and at the top of the nose), and is associated with the presence of a unibrow.[28]
Germline mutations in disease
Germline mutations of the Pax3 gene cause the splotch phenotype in mice.[29][30] At the molecular level, this phenotype is caused by point mutations or deletions that alter or abolish Pax3 transcriptional function. In the heterozygous state, the splotch phenotype is characterized by white patches in the belly, tail and feet. These white spots are attributed to localized deficiencies in pigment-forming melanocytes resulting from neural crest cell defects. In the homozygous state, these Pax3 mutations cause embryonic lethality, which is associated with prominent neural tube closure defects and abnormalities of neural crest-derived structures, such as melanocytes, dorsal root ganglia and enteric ganglia. Heart malformations also result from the loss of cardiac neural crest cells, which normally contribute to the cardiac outflow tract and innervation of the heart. Finally, limb musculature does not develop in the homozygotes and axial musculature demonstrates varying abnormalities. These myogenic effects are caused by increased cell death of myogenic precursors in the dermomyotome and diminished migration from the dermomyotome.
Germline mutations of the PAX3 gene occur in the human disease Waardenburg syndrome,[31] which consists of four autosomal dominant genetic disorders (WS1, WS2, WS3 and WS4).[32] Of the four subtypes, WS1 and WS3 are usually caused by PAX3 mutations. All four subtypes are characterized by hearing loss, eye abnormalities and pigmentation disorders. In addition, WS1 is frequently associated with a midfacial alteration called dystopia canthorum, while WS3 (Klein-Waardenburg syndrome) is frequently distinguished by musculoskeletal abnormalities affecting the upper limbs. Most WS1 cases are caused by heterozygous PAX3 mutations while WS3 is caused by either partial or total deletion of PAX3 and contiguous genes or by smaller PAX3 mutations in the heterozygous or homozygous state. These PAX3 mutations in WS1 and WS3 include missense, nonsense and splicing mutations; small insertions; and small or gross deletions. Though these changes are usually not recurrent, the mutations generally occur in exons 2 through 6 with exon 2 mutations being most common. As these exons encode the paired box and homeodomain, these mutations often affect DNA binding function.
Mutations in human cancer
Alveolar rhabdomyosarcoma (ARMS) is an aggressive soft tissue sarcoma that occurs in children and is usually characterized by a recurrent t(2;13)(q35;q14) chromosomal translocation.[33][34] This 2;13 translocation breaks and rejoins portions of the PAX3 and FOXO1 genes to generate a PAX3-FOXO1 fusion gene that expresses a PAX3-FOXO1 fusion transcript encoding a PAX3-FOXO1 fusion protein.[35] PAX3 and FOXO1 encode transcription factors, and the translocation results in a fusion transcription factor containing the N-terminal PAX3 DNA-binding domain and the C-terminal FOXO1 transactivation domain. A smaller subset of ARMS cases is associated with less common fusions of PAX7 to FOXO1 or rare fusions of PAX3 to other transcription factors, such as NCOA1.[36][37] Compared to the wild-type PAX3 protein, the PAX3-FOXO1 fusion protein more potently activates PAX3 target genes.[13] In ARMS cells, PAX3-FOXO1 usually functions as a transcriptional activator and excessively increases expression of downstream target genes.[38][39] In addition, PAX3-FOXO1 binds along with MYOD1, MYOG and MYCN as well as chromatin structural proteins, such as CHD4 and BRD4, to contribute to the formation of super enhancers in the vicinity of a subset of these target genes.[40] These dysregulated target genes contribute to tumorigenesis by altering signaling pathways that affect proliferation, cell death, myogenic differentiation, and migration.
A t(2;4)(q35;q31.1) chromosomal translocation that fuses the PAX3 and MAML3 genes occurs in biphenotypic sinonasal sarcoma (BSNS), a low-grade adult malignancy associated with both myogenic and neural differentiation.[41] MAML3 encodes a transcriptional coactivator involved in Notch signaling. The PAX3-MAML3 fusion juxtaposes the N-terminal PAX3 DNA binding domain with the C-terminal MAML3 transactivation domain to create another potent activator of target genes with PAX3 binding sites. Of note, PAX3 is rearranged without MAML3 involvement in a smaller subset of BSNS cases, and some of these variant cases contain a PAX3-NCOA1 or PAX3-FOXO1 fusion.[42][43] Though PAX3-FOXO1 and PAX3-NCOA1 fusions can be formed in both ARMS and BSNS, there are differences in the pattern of activated downstream target genes suggesting that the cell environment has an important role in modulating the output of these fusion transcription factors.
In addition to tumors with PAX3-related fusion genes, there are several other tumor categories that express the wild-type PAX3 gene. The presence of PAX3 expression in some tumors can be explained by their derivation from developmental lineages normally expressing wild-type PAX3. For example, PAX3 is expressed in cancers associated with neural tube-derived lineages, (e.g., glioblastoma), neural crest-derived lineages (e.g., melanoma) and myogenic lineages (e.g., embryonal rhabdomyosarcoma).[44][45][46][47] However, PAX3 is also expressed in other cancer types without a clear relationship to a PAX3-expressing developmental lineages, such as breast carcinoma and osteosarcoma.[48][49] In these wild-type PAX3-expressing cancers, PAX3 function impacts on the control of proliferation, apoptosis, differentiation and motility.[44][45] Therefore, wild-type PAX3 exerts a regulatory role in tumorigenesis and tumor progression, which may be related to its role in normal development.
Notes
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000135903 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000004872 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ Stuart ET, Kioussi C, Gruss P (1994). "Mammalian Pax genes". review. Annual Review of Genetics. 28: 219–36. doi:10.1146/annurev.ge.28.120194.001251. PMID 7893124.
- ↑ Wang Q, Fang WH, Krupinski J, Kumar S, Slevin M, Kumar P (December 2008). "Pax genes in embryogenesis and oncogenesis". review. Journal of Cellular and Molecular Medicine. 12 (6A): 2281–94. doi:10.1111/j.1582-4934.2008.00427.x. PMC 4514106. PMID 18627422.
- ↑ Vogan KJ, Underhill DA, Gros P (December 1996). "An alternative splicing event in the Pax-3 paired domain identifies the linker region as a key determinant of paired domain DNA-binding activity". primary. Molecular and Cellular Biology. 16 (12): 6677–86. doi:10.1128/mcb.16.12.6677. PMC 231670. PMID 8943322.
- ↑ Baldwin CT, Hoth CF, Macina RA, Milunsky A (August 1995). "Mutations in PAX3 that cause Waardenburg syndrome type I: ten new mutations and review of the literature". review. American Journal of Medical Genetics. 58 (2): 115–22. doi:10.1002/ajmg.1320580205. PMID 8533800.
- ↑ Jun S, Desplan C (September 1996). "Cooperative interactions between paired domain and homeodomain". primary. Development. 122 (9): 2639–50. doi:10.1242/dev.122.9.2639. PMID 8787739.
- ↑ Wilson D, Sheng G, Lecuit T, Dostatni N, Desplan C (November 1993). "Cooperative dimerization of paired class homeo domains on DNA". primary. Genes & Development. 7 (11): 2120–34. doi:10.1101/gad.7.11.2120. PMID 7901121.
- ↑ Phelan SA, Loeken MR (July 1998). "Identification of a new binding motif for the paired domain of Pax-3 and unusual characteristics of spacing of bipartite recognition elements on binding and transcription activation". primary. The Journal of Biological Chemistry. 273 (30): 19153–9. doi:10.1074/jbc.273.30.19153. PMID 9668101.
- ↑ Bennicelli JL, Fredericks WJ, Wilson RB, Rauscher FJ, Barr FG (July 1995). "Wild type PAX3 protein and the PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma contain potent, structurally distinct transcriptional activation domains". primary. Oncogene. 11 (1): 119–30. PMID 7624119.
- 1 2 Bennicelli JL, Edwards RH, Barr FG (May 1996). "Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma". primary. Proceedings of the National Academy of Sciences of the United States of America. 93 (11): 5455–9. Bibcode:1996PNAS...93.5455B. doi:10.1073/pnas.93.11.5455. PMC 39267. PMID 8643596.
- ↑ Mayanil CS, George D, Freilich L, Miljan EJ, Mania-Farnell B, McLone DG, Bremer EG (December 2001). "Microarray analysis detects novel Pax3 downstream target genes". primary. The Journal of Biological Chemistry. 276 (52): 49299–309. doi:10.1074/jbc.M107933200. PMID 11590174.
- ↑ Soleimani VD, Punch VG, Kawabe Y, Jones AE, Palidwor GA, Porter CJ, Cross JW, Carvajal JJ, Kockx CE, van IJcken WF, Perkins TJ, Rigby PW, Grosveld F, Rudnicki MA (June 2012). "Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs". primary. Developmental Cell. 22 (6): 1208–20. doi:10.1016/j.devcel.2012.03.014. PMC 3376216. PMID 22609161.
- ↑ Schäfer BW, Czerny T, Bernasconi M, Genini M, Busslinger M (November 1994). "Molecular cloning and characterization of a human PAX-7 cDNA expressed in normal and neoplastic myocytes". primary. Nucleic Acids Research. 22 (22): 4574–82. doi:10.1093/nar/22.22.4574. PMC 308503. PMID 7527137.
- ↑ Lang D, Epstein JA (April 2003). "Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer". primary. Human Molecular Genetics. 12 (8): 937–45. doi:10.1093/hmg/ddg107. PMID 12668617.
- ↑ Diao Y, Guo X, Li Y, Sun K, Lu L, Jiang L, Fu X, Zhu H, Sun H, Wang H, Wu Z (August 2012). "Pax3/7BP is a Pax7- and Pax3-binding protein that regulates the proliferation of muscle precursor cells by an epigenetic mechanism". primary. Cell Stem Cell. 11 (2): 231–41. doi:10.1016/j.stem.2012.05.022. PMID 22862948.
- ↑ Hollenbach AD, McPherson CJ, Lagutina I, Grosveld G (April 2002). "The EF-hand calcium-binding protein calmyrin inhibits the transcriptional and DNA-binding activity of Pax3". primary. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression. 1574 (3): 321–8. doi:10.1016/s0167-4781(02)00230-0. PMID 11997098.
- ↑ Dietz KN, Miller PJ, Iyengar AS, Loupe JM, Hollenbach AD (June 2011). "Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation". primary. The International Journal of Biochemistry & Cell Biology. 43 (6): 936–45. doi:10.1016/j.biocel.2011.03.010. PMC 3095663. PMID 21440083.
- ↑ Kubic JD, Mascarenhas JB, Iizuka T, Wolfgeher D, Lang D (August 2012). "GSK-3 promotes cell survival, growth, and PAX3 levels in human melanoma cells". primary. Molecular Cancer Research. 10 (8): 1065–76. doi:10.1158/1541-7786.MCR-11-0387. PMC 3422428. PMID 22679108.
- ↑ Boutet SC, Disatnik MH, Chan LS, Iori K, Rando TA (July 2007). "Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors". primary. Cell. 130 (2): 349–62. doi:10.1016/j.cell.2007.05.044. PMID 17662948.
- ↑ Ichi S, Boshnjaku V, Shen YW, Mania-Farnell B, Ahlgren S, Sapru S, Mansukhani N, McLone DG, Tomita T, Mayanil CS (February 2011). "Role of Pax3 acetylation in the regulation of Hes1 and Neurog2". primary. Molecular Biology of the Cell. 22 (4): 503–12. doi:10.1091/mbc.E10-06-0541. PMC 3038648. PMID 21169561.
- ↑ Buckingham M, Relaix F (2007). "The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions". review. Annual Review of Cell and Developmental Biology. 23: 645–73. doi:10.1146/annurev.cellbio.23.090506.123438. PMID 17506689.
- ↑ Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P (May 1991). "Pax-3, a novel murine DNA binding protein expressed during early neurogenesis". primary. The EMBO Journal. 10 (5): 1135–47. doi:10.1002/j.1460-2075.1991.tb08054.x. PMC 452767. PMID 2022185.
- ↑ Monsoro-Burq AH (August 2015). "PAX transcription factors in neural crest development". review. Seminars in Cell & Developmental Biology. 44: 87–96. doi:10.1016/j.semcdb.2015.09.015. PMID 26410165.
- ↑ Wu M, Li J, Engleka KA, Zhou B, Lu MM, Plotkin JB, Epstein JA (June 2008). "Persistent expression of Pax3 in the neural crest causes cleft palate and defective osteogenesis in mice". primary. The Journal of Clinical Investigation. 118 (6): 2076–87. doi:10.1172/JCI33715. PMC 2381747. PMID 18483623.
- ↑ Adhikari, Kaustubh. "How we discovered the genetic origin of the 'monobrow' and other hair traits". The Conversation. Retrieved 2018-08-27.
- ↑ Epstein DJ, Vekemans M, Gros P (November 1991). "Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3". primary. Cell. 67 (4): 767–74. doi:10.1016/0092-8674(91)90071-6. PMID 1682057. S2CID 25966319.
- ↑ Tremblay P, Gruss P (1994). "Pax: genes for mice and men". review. Pharmacology & Therapeutics. 61 (1–2): 205–26. doi:10.1016/0163-7258(94)90063-9. PMID 7938171.
- ↑ Baldwin CT, Hoth CF, Amos JA, da-Silva EO, Milunsky A (February 1992). "An exonic mutation in the HuP2 paired domain gene causes Waardenburg's syndrome". primary. Nature. 355 (6361): 637–8. Bibcode:1992Natur.355..637B. doi:10.1038/355637a0. PMID 1347149. S2CID 4319436.
- ↑ Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N (April 2010). "Review and update of mutations causing Waardenburg syndrome". review. Human Mutation. 31 (4): 391–406. doi:10.1002/humu.21211. PMID 20127975. S2CID 12278025.
- ↑ Barr FG (September 2001). "Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma". review. Oncogene. 20 (40): 5736–46. doi:10.1038/sj.onc.1204599. PMID 11607823.
- ↑ Arndt, C. A.; Crist, W. M. (1999-07-29). "Common musculoskeletal tumors of childhood and adolescence". The New England Journal of Medicine. 341 (5): 342–352. doi:10.1056/NEJM199907293410507. ISSN 0028-4793. PMID 10423470.
- ↑ Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, Emanuel BS, Rovera G, Barr FG (November 1993). "Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma". primary. Nature Genetics. 5 (3): 230–5. doi:10.1038/ng1193-230. PMID 8275086. S2CID 12374322.
- ↑ Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. (February 2014). "Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors". primary. Cancer Discovery. 4 (2): 216–31. doi:10.1158/2159-8290.CD-13-0639. PMC 4462130. PMID 24436047.
- ↑ Barr, Frederic G. (1995-02-15). "Molecular Assays for Chromosomal Translocations in the Diagnosis of Pediatric Soft Tissue Sarcomas". JAMA: The Journal of the American Medical Association. 273 (7): 553–7. doi:10.1001/jama.1995.03520310051029. ISSN 0098-7484. PMID 7530783.
- ↑ Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, Anderson MJ (July 2006). "Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas". primary. Cancer Research. 66 (14): 6936–46. doi:10.1158/0008-5472.CAN-05-4578. PMID 16849537.
- ↑ Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, Yang F, Pineda M, Helman LJ, Meltzer PS (August 2010). "Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer". primary. Cancer Research. 70 (16): 6497–508. doi:10.1158/0008-5472.CAN-10-0582. PMC 2922412. PMID 20663909.
- ↑ Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, et al. (August 2017). "PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability". primary. Cancer Discovery. 7 (8): 884–899. doi:10.1158/2159-8290.CD-16-1297. PMC 7802885. PMID 28446439.
- ↑ Wang X, Bledsoe KL, Graham RP, Asmann YW, Viswanatha DS, Lewis JE, Lewis JT, Chou MM, Yaszemski MJ, Jen J, Westendorf JJ, Oliveira AM (July 2014). "Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma". primary. Nature Genetics. 46 (7): 666–8. doi:10.1038/ng.2989. PMC 4236026. PMID 24859338.
- ↑ Fritchie KJ, Jin L, Wang X, Graham RP, Torbenson MS, Lewis JE, et al. (December 2016). "Fusion gene profile of biphenotypic sinonasal sarcoma: an analysis of 44 cases". primary. Histopathology. 69 (6): 930–936. doi:10.1111/his.13045. PMID 27454570. S2CID 9521589.
- ↑ Huang SC, Ghossein RA, Bishop JA, Zhang L, Chen TC, Huang HY, Antonescu CR (January 2016). "Novel PAX3-NCOA1 Fusions in Biphenotypic Sinonasal Sarcoma With Focal Rhabdomyoblastic Differentiation". The American Journal of Surgical Pathology. 40 (1): 51–9. doi:10.1097/PAS.0000000000000492. PMC 4679641. PMID 26371783.
- 1 2 Xia L, Huang Q, Nie D, Shi J, Gong M, Wu B, Gong P, Zhao L, Zuo H, Ju S, Chen J, Shi W (July 2013). "PAX3 is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells". primary. Brain Research. 1521: 68–78. doi:10.1016/j.brainres.2013.05.021. PMID 23701726. S2CID 11206561.
- 1 2 Scholl FA, Kamarashev J, Murmann OV, Geertsen R, Dummer R, Schäfer BW (February 2001). "PAX3 is expressed in human melanomas and contributes to tumor cell survival". primary. Cancer Research. 61 (3): 823–6. PMID 11221862.
- ↑ Barr FG, Fitzgerald JC, Ginsberg JP, Vanella ML, Davis RJ, Bennicelli JL (November 1999). "Predominant expression of alternative PAX3 and PAX7 forms in myogenic and neural tumor cell lines". primary. Cancer Research. 59 (21): 5443–8. PMID 10554014.
- ↑ Plummer RS, Shea CR, Nelson M, Powell SK, Freeman DM, Dan CP, Lang D (May 2008). "PAX3 expression in primary melanomas and nevi". Modern Pathology. 21 (5): 525–30. doi:10.1038/modpathol.3801019. PMC 2987639. PMID 18327212.
- ↑ Jones AM, Mitter R, Poulsom R, Gillett C, Hanby AM, Tomlinson IP, Sawyer EJ (December 2008). "mRNA expression profiling of phyllodes tumours of the breast: identification of genes important in the development of borderline and malignant phyllodes tumours". primary. The Journal of Pathology. 216 (4): 408–17. doi:10.1002/path.2439. PMID 18937276. S2CID 5294598.
- ↑ Liu Q, Yang G, Qian Y (April 2017). "Loss of MicroRNA-489-3p promotes osteosarcoma metastasis by activating PAX3-MET pathway". primary. Molecular Carcinogenesis. 56 (4): 1312–1321. doi:10.1002/mc.22593. PMID 27859625. S2CID 4439256.
Further reading
- Wachtel M, Schäfer BW (2017). "PAX3-FOXO1: Zooming in on an "undruggable" target". review. Seminars in Cancer Biology. 50: 115–123. doi:10.1016/j.semcancer.2017.11.006. PMID 29146205.
- Kubo T, Shimose S, Fujimori J, Furuta T, Ochi M (2015). "Prognostic value of PAX3/7-FOXO1 fusion status in alveolar rhabdomyosarcoma: Systematic review and meta-analysis". review. Critical Reviews in Oncology/Hematology. 96 (1): 46–53. doi:10.1016/j.critrevonc.2015.04.012. PMID 26008753.
- Buckingham M, Relaix F (2015). "PAX3 and PAX7 as upstream regulators of myogenesis". review. Seminars in Cell & Developmental Biology. 44: 115–25. doi:10.1016/j.semcdb.2015.09.017. PMID 26424495.
- Eccles MR, He S, Ahn A, Slobbe LJ, Jeffs AR, Yoon HS, Baguley BC (2013). "MITF and PAX3 Play Distinct Roles in Melanoma Cell Migration; Outline of a "Genetic Switch" Theory Involving MITF and PAX3 in Proliferative and Invasive Phenotypes of Melanoma". review. Frontiers in Oncology. 3: 229. doi:10.3389/fonc.2013.00229. PMC 3769631. PMID 24062982.
- Olanich ME, Barr FG (2013). "A call to ARMS: targeting the PAX3-FOXO1 gene in alveolar rhabdomyosarcoma". review. Expert Opinion on Therapeutic Targets. 17 (5): 607–23. doi:10.1517/14728222.2013.772136. PMC 6699745. PMID 23432728.
- Medic S, Ziman M (2009). "PAX3 across the spectrum: from melanoblast to melanoma". review. Critical Reviews in Biochemistry and Molecular Biology. 44 (2–3): 85–97. doi:10.1080/10409230902755056. PMID 19401874. S2CID 20634846.
- Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D (2008). "Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease". review. Pigment Cell & Melanoma Research. 21 (6): 627–45. doi:10.1111/j.1755-148X.2008.00514.x. PMC 2979299. PMID 18983540.
External links
- PAX3+protein,+human at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- Overview of all the structural information available in the PDB for UniProt: P23760 (Paired box protein Pax-3) at the PDBe-KB.