藥物設計
藥物設計(英語:),常稱理性藥物設計(Rational drug design),是根據生物靶点(Biological target)的現有知識尋找與發现新型藥物的過程[1]。最常见的药物类型如有机小分子药物,可激活或抑制蛋白质等生物分子功能,进而为患者在治疗中获益。药物设计可狭义地定义为药物分子设计,这些药物分子的形状和原子所带电荷与生物分子靶标存在互补关系,即“锁钥模型”(Lock and key model),因此药物分子会与生物靶标存在结合力。使用電腦分子建構技術進行藥物設計可稱為计算机輔助藥物設計(Computer-aided drug design,CADD)。根據對於生物目標的化學結構來進行設計,稱為結構藥物設計(Structure-based drug design)[2]。除了小分子之外,现今包括多肽与單克隆抗體治療在内的众多生物治療方式,在制药领域变得越发重要。与此同时,各种提高药物亲和力、选择性和稳定性的算法在基于蛋白质治疗的领域中也持续发展[3]。
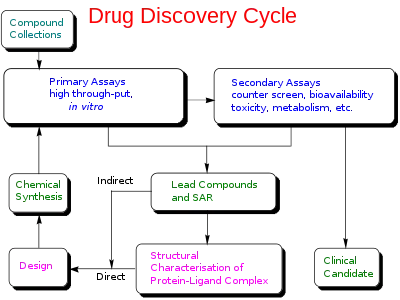
“药物设计”的表述在某种意义上并不准确。更准确应是配体设计,即设计某分子使其与目标靶标紧密结合[4]。尽管现有技术对于预测结合亲和力相当成功,但在配体分子成为安全有效的药物之前须对该分子的众多成药因素进行优化,如:生物利用度、代谢半衰期、药物副作用等,然而以上特性基于现有技术通常难以预测。由于药物设计的高失败率,尤其在药物开发的临床阶段存在高失败率(尤其在临床II期[5]),在药物设计过程的早期,药物化学家通常将更多精力集中在选择出更优的候选药物上。这些候选药物的物理化学性质会基于避免药物不良反应进行考量,从而提高候选药物推向获批上市的可能性[6]。此外,药物体外实验与计算辅助设计相辅相成,越来越多地用于早期药物发现阶段,以挑选出更优ADME(吸收、分布、代谢和排泄)性质和毒理学性质的化合物[7]。
药物靶点
生物分子靶标如最常见的蛋白质或核酸,是参与特定代谢或信号通路的关键分子,该通路与特定疾病的病理学成因或微生物病原体的传染性相关。一个潜在的药物靶点不一定直接致病,而应与改善或影响疾病相关联[8]。在某些情况下,小分子被设计用于增强或抑制特定疾病中修饰途径的目标功能。诸如受体激动剂、拮抗剂、反向激动剂或调节剂;酶激活剂或抑制剂;或离子通道开放剂和阻滞剂[9]的小分子药物被设计为与靶标结合位点通过结合力互补[10]。由于药物分子与脱靶分子的相互作用可能会导致不良反应,小分子药物可以设计为不影响任何其他重要“脱靶”分子(通常称为抗靶标)[11]。由于靶标结合位点存在相似性,通过序列同源性鉴定的存在密切关联的多个靶标与设计的化合物可能均存在反应性或结合力,从而导致潜在的不良反应。
通过化学合成的有机小分子是现有药物发现中的一大类,而通过生物过程生产的生物药物在制药领域正变得越来越广泛[12]。此外,基于mRNA的基因沉默技术也同时越来越多地进入新的治疗领域[13]。
合理的药物发现
传统的药物发现方法(亦称为正向药理学),是基于不同化合物与培养细胞或动物进行反复试验,通过匹配化合物结构与药效(Structure-activity relationship,SAR)以寻找候选药物。与之相区别的,合理的药物设计(亦称为反向药理学)初始于假定调节某生物靶标可具有治疗价值。选择某生物分子作为药物靶标需要两个基本要素:其一有证据表明调节该靶标可缓解病症。如研究发现表明:生物靶标的突变与某些疾病之间存在关联性[14];其二该靶标是“可成药”的,即靶标可结合小分子,并可通过小分子的活性进行调控[15]。
一旦确定了合适的靶标,通常可通过克隆、生产和纯化手段合成靶标分子。将纯化的蛋白质靶标用于建立药物筛选试验平台,并确定靶标的三维结构。
此时若存在化合物库资源,通常可通过化合物库筛选一轮潜在可与靶标结合的小分子,也称为高通量筛选。此外若靶点为确定结构的蛋白质,还可对候选化合物进行计算机虚拟筛选。理想情况下,候选药物化合物还能同时具备“ADME相关类药性”,包括满足口服生物利用度、化学稳定性、代谢稳定性及足够低的毒副作用的ADME成药性要求[16]。有诸多方法可评估类药性,如里宾斯基五规则和其他评估手段如亲脂性等[17]。此外众多药物化学家也提出了许多预测类药性的方法[18][19]。
计算机辅助药物设计
药物设计的最初目标是预测某分子是否会与目标靶标结合,并预测其结合强度强弱。通常基于分子力学或分子动力学估算小分子与其生物靶标之间的分子-分子间相互作用强度。该方法还可用于预测小分子的空间构象,并模拟小分子与之结合时导致的靶标构象变化[20][21]。半经验的、从头算量子化学方法或密度泛函理论通常是分子力学计算优化参数的主要手段,并可估算候选药物的电子特性如:静电势、极化率等),从而影响药物-靶标亲和力[22]。
分子力学方法也可提供靶标结合亲和力的半定量预测。此外,基于药物设计知识的评分函数是估算结合亲和力的另一种主流方式。这些评分函数使用线性回归、机器学习、神经网络或其他统计技术,通过将实验测定的亲和力进行拟合可以推算小分子与靶标之间的相互作用能量,并将数据整合以推导出可预测结合亲和力方程[23] [24]。
理想情况下,计算机辅助设计可预测合成化合物的亲和力,因此理论上只需要合成一类化合物便可预测其成药可能性,从而节省药物研发中大量的时间和成本。而实际药物研发过程中,计算机辅助设计并不能解决所有问题,通常充其量只可定性地估算亲和力。因此在现今的药物研发过程中,发现较优的药物分子前要进行多轮的设计、合成及测试并反复迭代。计算机辅助药物设计可通过减少所需的迭代次数以加速药物发现过程,并提供一些新颖的化学结构[25] [26]。
基于计算机辅助药物设计可用于药物发现的以下任何阶段:
- 基于结构或配体的设计使用计算机虚拟筛选以寻找苗头化合物(Hit compound)
- 基于结构的药物设计、QSAR等知识,对于苗头化合物至先导化合物(Lead compound)的亲和力与选择性进行优化
- 在维持与配体亲和力条件下,对于先导化合物的成药性或ADME性质进行优化
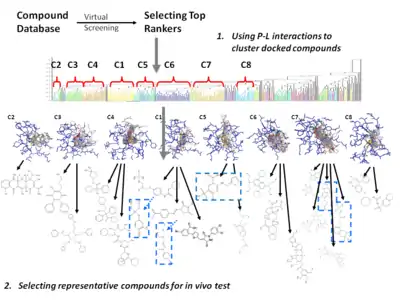
为了解决评分函数计算中结合亲和力预测的不准确性,通常可进行蛋白质-配体相互作用与化合物三维结构信息进行分析和佐证。对于基于结构的药物设计,已经开发出几种侧重于蛋白质-配体相互作用的筛选后分析方法,以提高发现潜在候选药物的效率:
药物设计策略类型
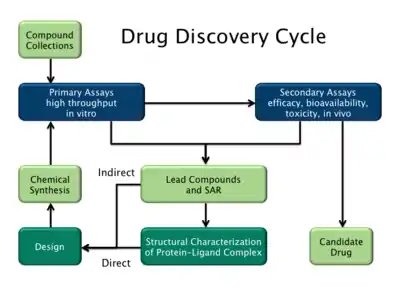
现在存在两种主流药物设计策略:第一种称为基于配体(Ligand)的药物设计,第二种称为基于结构的药物设计[31]。
基于配体的药物设计
基于配体的药物设计(或间接药物设计)基于与已知生物靶标与确定分子结合的数据。这些分子可用于推测药效团模型,即分子中可结合靶标中最小且必要的结构片段[32]。同样的,构建生物靶标模型可基于与其结合的分子,同时该建模反之可用于设计与靶标相互作用的新化学实体(New chemical entity,NCE)。又或者,该建模还可归纳量化结构-活性关系(QSAR),即通过计算分子性质与实验测定的生物活性之间的关联度。这些QSAR关系还可继续通过计算机模拟迭代,以预测新的候选类似物的生物活性[33]。
基于结构的药物设计
基于结构的药物设计(或直接药物设计)可通过X 射线晶体学或核磁共振光谱学等方法获得的生物靶标的三维结构数据[34]。若靶标三维空间结构无法被测定,则可根据相关蛋白质的实验测定结构数据,以创建靶标的同源模型。通过生物靶标结构,可以使用化合物配体-靶标相互作用模型和药物化学家的直觉预测并设计高亲和力和选择性结合靶标的候选药物。又或者可以基于各种自动运算程序推选新的候选药物[35]。
当前基于结构的药物设计方法大致可分为三大类[36]:第一种方法基于小分子三维结构大型数据库以寻找可与受体结合的新配体分子,通过使用计算机快速对接(Docking)程序找到适合受体结合口袋(Binding pocket)的配体分子。这类方法称为虚拟筛选。第二类方法是通过新配体进行从头设计。在这类方法中,通过计算机模拟组装小分子片段并在结合口袋的空间约束下逐步构建形成整个配体分子,这些化学小分子片段可以是单个原子或分子结构碎片。这种方法的优点是可不被任何化学数据库中的结构所束缚,可进行较自由的结构创造[37][38][39]。第三类方法是通过评价结合腔内拟定的类似物,对已知配体进行不断优化[36]。
识别结合位点
基于结构的药物设计的第一个步骤是识别结合位点[40][41]。结合位点的识别通常取决于靶标蛋白质上可结合小分子(配体分子)的凹面处,这些凹面位置可容纳与靶点结合所匹配大小的药物分子,而这些结合力强的分子也具有与配体结合的适当“热点(Hot spots)”如:疏水性表面结构、氢键结合位点等[40] [41]。
评分函数
基于结构的药物设计通过应用分子识别原理,以蛋白质结构作为设计新配体的基础。药物设计中通常倾向选择与靶标选择性地高亲和力结合的分子,从而使得药物的效价更高而副作用更低。因此设计并获得潜在新配体的最重要原则之一是预测特定配体化合物与其靶标(或已知反靶标)的结合亲和力,并将该亲和力作为关键的候选药物选择标准。 [42]
Böhm开发了一种可早期用于通用经验的评分函数,该函数可用于预测配体与受体的结合能[43][44]。该经验评分函数如下:
其中:

- ΔG 0 – 经验推导的结合能偏移,部分对应结合配体的平移和旋转熵的总体损失的结合能
- ΔG hb – 氢键贡献的结合能
- ΔG ionic – 离子相互作用贡献的结合能
- ΔG lip – 亲脂性相互作用贡献的结合能,其中 |Alipo |代表配体与受体之间亲脂性接触的表面积
- ΔG rot – 配体结合时冻结(Freeze)可旋转配体键而导致的熵罚(Entropy penalty)
更为通用的热力学“主方程”如下: [45]
![{\displaystyle {\begin{array}{lll}\Delta G_{\text{bind}}=-RT\ln K_{\text{d}}\\[1.3ex]K_{\text{d}}={\dfrac {[{\text{Ligand}}][{\text{Receptor}}]}{[{\text{Complex}}]}}\\[1.3ex]\Delta G_{\text{bind}}=\Delta G_{\text{desolvation}}+\Delta G_{\text{motion}}+\Delta G_{\text{configuration}}+\Delta G_{\text{interaction}}\end{array}}}](../I/ba49ddd9dec7415d129787213744ca1afcd2d021.svg)
- desolvation(去溶剂化)——将配体去溶剂化的的焓罚
- motion(运动)——当配体与受体结合时减少一定自由度的熵罚
- configuration (构象)——将配体置于其“活性”构象所需的构象张力
- interaction (相互作用)——配体与其受体发生溶剂化导致的焓增
以上的评分函数将整体结合自由能分解为已知的对结合过程很重要的独立分量。每个分量均反映配体与其靶受体结合过程中某种自由能的变化。主方程是这些分量的线性组合。根据吉布斯自由能方程,建立解离平衡常数Kd与自由能分量之间的关系。
不同的计算方法可用于估计主方程的每个分量。例如,配体结合的极性表面积变化可用于估算去溶剂化分量;配体结合时冻结可旋转键的数量与运动分量成正比;基于分子力学计算可估算构象或张力得分量;基于非极性表面的变化、形成氢键数等方法可估算相互作用分量。在实际的药物发开过程中,主方程的各个分量部分使用多元线性回归拟合实际的实验测试数据。这可以通过包括多个配体和受体的多样化训练集进行,以生成准确性较低但更通用于“全局设计”的模型,或者使用单一且更受限的配体与受体生成相较更准确却通用性较低的“局部设计”模型 [46]。
案例
合理药物设计中具体个案通常使用从X射线晶体学和核磁共振光谱学等技术获取有关生物分子的三维信息。计算机辅助药物设计对于高分辨率的已知目标蛋白结构与活性配体的结合处理起来得心应手,这种药物发现策略被称为基于结构的药物设计。
第一个相关案例是碳酸酐酶抑制剂多佐胺应用基于结构的药物设计发现,并于1995年获得当局批准[47][48]。
另一个重要案例研究是伊马替尼,该药物是一种酪氨酸激酶抑制剂,专门针对费城染色体阳性白血病(慢性粒细胞性白血病和偶发性急性淋巴细胞性白血病)特有的bcr-abl融合蛋白进行药物设计。伊马替尼与以往传统的癌症药物存在本质性差异:主流传统化疗药物只针对快速分裂的细胞起作用,而无法区分癌细胞和正常组织,而伊马替尼可选择性针对癌症细胞作用以减少药品不良反应[49]。
更多案例包括:
- 5-HT3拮抗剂
- 乙酰胆碱受体激动剂
- 血管紧张素受体激动剂
- Bcr-Abl蛋白酪氨酸激酶抑制剂
- 大麻素受体拮抗剂
- CCR5受体拮抗剂
- 环氧化酶2抑制剂
- 二肽基肽酶-4抑制剂
- HIV蛋白酶抑制剂
- NK1受体拮抗剂
- 非核苷类反转录酶抑制剂
- 核苷类反转录酶抑制剂
- PDE5抑制剂
- 质子泵抑制剂
- 肾素抑制剂
- 曲坦类抑制剂
- TRPV1拮抗剂
- c-Met抑制剂
评论
- 在制药领域中也有不少学者认为高度刚性和同质化的合理药物设计虽然提高了药品开发的速度并减少了药物设计失败的可能性,却同时抑制了药物发现的偶然性[50]。
参考资料
- Madsen U, Krogsgaard-Larsen P, Liljefors T. . Washington, DC: Taylor & Francis. 2002. ISBN 0-415-28288-8.
- Reynolds CH, Merz KM, Ringe D (编). 1. Cambridge, UK: Cambridge University Press. 2010.
- Shirai H, Prades C, Vita R, Marcatili P, Popovic B, Xu J, Overington JP, Hirayama K, Soga S, Tsunoyama K, Clark D, Lefranc MP, Ikeda K. . Biochimica et Biophysica Acta. Nov 2014, 1844 (11): 2002–2015. PMID 25110827. doi:10.1016/j.bbapap.2014.07.006.
- Tollenaere JP. . Pharmacy World & Science. Apr 1996, 18 (2): 56–62. PMID 8739258. S2CID 21550508. doi:10.1007/BF00579706.
- . BIO, Informa Pharma Intelligence, and QLS Advisors. Feb 2021 [2023-06-20]. (原始内容存档于2023-02-08).
- Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, Wallace O, Weir A. . Nature Reviews Drug Discovery. 2015, 14 (7): 475–86. PMID 26091267. S2CID 25292436. doi:10.1038/nrd4609.
- Yu H, Adedoyin A. . Drug Discovery Today. Sep 2003, 8 (18): 852–61. PMID 12963322. doi:10.1016/S1359-6446(03)02828-9.
- Dixon SJ, Stockwell BR. . Current Opinion in Chemical Biology. Dec 2009, 13 (5–6): 549–55. PMC 2787993
 . PMID 19740696. doi:10.1016/j.cbpa.2009.08.003.
. PMID 19740696. doi:10.1016/j.cbpa.2009.08.003. - Imming P, Sinning C, Meyer A. . Nature Reviews. Drug Discovery. Oct 2006, 5 (10): 821–34. PMID 17016423. S2CID 8872470. doi:10.1038/nrd2132.
- Anderson AC. . Chemistry & Biology. Sep 2003, 10 (9): 787–97. PMID 14522049. doi:10.1016/j.chembiol.2003.09.002 .
- Recanatini M, Bottegoni G, Cavalli A. . Drug Discovery Today: Technologies. Dec 2004, 1 (3): 209–15. PMID 24981487. doi:10.1016/j.ddtec.2004.10.004.
- Wu-Pong S, Rojanasakul Y. 2nd. Totowa, NJ Humana Press: Humana Press. 2008. ISBN 978-1-59745-532-9.
- Scomparin A, Polyak D, Krivitsky A, Satchi-Fainaro R. . Biotechnology Advances. Apr 2015, 33 (6): 1294–309. PMID 25916823. doi:10.1016/j.biotechadv.2015.04.008.
- Ganellin CR, Jefferis R, Roberts SM. . Elsevier. 2013. ISBN 9780123971760.
- Yuan Y, Pei J, Lai L. . Current Pharmaceutical Design. Dec 2013, 19 (12): 2326–33. PMID 23082974. doi:10.2174/1381612811319120019.
- Rishton GM. . Drug Discovery Today. Jan 2003, 8 (2): 86–96. PMID 12565011. doi:10.1016/s1359644602025722.
- Hopkins AL. Wermuth CG , 编. 3. Academic Press. 2011: 521–527. ISBN 978-0-12-374194-3.
- Kirchmair J. . Wiley's Methods and Principles in Medicinal Chemistry 63. Wiley-VCH. 2014. ISBN 978-3-527-67301-8.
- Ethiraj SK, Levinthal D. . Administrative Science Quarterly (Sage Publications, Inc. on behalf of the Johnson Graduate School of Management, Cornell University). Sep 2004, 49 (3): 404–437 [2023-05-04]. JSTOR 4131441. S2CID 142910916. SSRN 604123 . doi:10.2307/4131441. (原始内容存档于2022-05-26).
- Fosgerau K, Hoffmann T. . Drug Discovery Today. January 2015, 20 (1): 122–128. PMID 25450771. doi:10.1016/j.drudis.2014.10.003 .Fosgerau K, Hoffmann T (January 2015). "Peptide therapeutics: current status and future directions". Drug Discovery Today. 20 (1): 122–128. doi:10.1016/j.drudis.2014.10.003. PMID 25450771 (页面存档备份,存于).
- Ciemny M, Kurcinski M, Kamel K, Kolinski A, Alam N, Schueler-Furman O, Kmiecik S. . Drug Discovery Today. August 2018, 23 (8): 1530–1537. PMID 29733895. doi:10.1016/j.drudis.2018.05.006 .Ciemny M, Kurcinski M, Kamel K, Kolinski A, Alam N, Schueler-Furman O, Kmiecik S (August 2018). "Protein-peptide docking: opportunities and challenges". Drug Discovery Today. 23 (8): 1530–1537. doi:10.1016/j.drudis.2018.05.006. PMID 29733895 (页面存档备份,存于).
- Lewis RA. . RSC Drug Discovery. Royal Society of Chemistry. 2011: 88–107. ISBN 978-1849731669. doi:10.1039/9781849733410-00088.
- Rajamani R, Good AC. . Current Opinion in Drug Discovery & Development. May 2007, 10 (3): 308–15. PMID 17554857.
- de Azevedo WF, Dias R. . Current Drug Targets. Dec 2008, 9 (12): 1031–9. PMID 19128212. doi:10.2174/138945008786949405.
- Singh J, Chuaqui CE, Boriack-Sjodin PA, Lee WC, Pontz T, Corbley MJ, Cheung HK, Arduini RM, Mead JN, Newman MN, Papadatos JL, Bowes S, Josiah S, Ling LE. . Bioorganic & Medicinal Chemistry Letters. Dec 2003, 13 (24): 4355–9. PMID 14643325. doi:10.1016/j.bmcl.2003.09.028.
- Becker OM, Dhanoa DS, Marantz Y, Chen D, Shacham S, Cheruku S, Heifetz A, Mohanty P, Fichman M, Sharadendu A, Nudelman R, Kauffman M, Noiman S. . Journal of Medicinal Chemistry. Jun 2006, 49 (11): 3116–35. PMID 16722631. doi:10.1021/jm0508641.
- Liang S, Meroueh SO, Wang G, Qiu C, Zhou Y. . Proteins. May 2009, 75 (2): 397–403. PMC 2656599 . PMID 18831053. doi:10.1002/prot.22252.
- Oda A, Tsuchida K, Takakura T, Yamaotsu N, Hirono S. . Journal of Chemical Information and Modeling. 2006, 46 (1): 380–91. PMID 16426072. doi:10.1021/ci050283k.
- Deng Z, Chuaqui C, Singh J. . Journal of Medicinal Chemistry. Jan 2004, 47 (2): 337–44. PMID 14711306. doi:10.1021/jm030331x.
- Amari S, Aizawa M, Zhang J, Fukuzawa K, Mochizuki Y, Iwasawa Y, Nakata K, Chuman H, Nakano T. . Journal of Chemical Information and Modeling. 2006, 46 (1): 221–30. PMID 16426058. doi:10.1021/ci050262q.
- 1. Cambridge, UK: Cambridge University Press. 2010. ISBN 978-0521887236.Reynolds CH, Merz KM, Ringe D, eds. (2010). Drug Design: Structure- and Ligand-Based Approaches (1 ed.). Cambridge, UK: Cambridge University Press. ISBN 978-0521887236.
- Guner OF. . La Jolla, Calif: International University Line. 2000. ISBN 978-0-9636817-6-8.
- Tropsha A. 1st. Cambridge, UK: Cambridge University Press. 2010: 151–164.
- Leach AR, Harren J. . Berlin: Springer. 2007. ISBN 978-1-4020-4406-9.
- Mauser H, Guba W. . Current Opinion in Drug Discovery & Development. May 2008, 11 (3): 365–74. PMID 18428090.
- Klebe G. . Journal of Molecular Medicine. 2000, 78 (5): 269–81. PMID 10954199. S2CID 21314020. doi:10.1007/s001090000084.
- Wang R, Gao Y, Lai L. . Journal of Molecular Modeling. 2000, 6 (7–8): 498–516. S2CID 59482623. doi:10.1007/s0089400060498.
- Schneider G, Fechner U. . Nature Reviews. Drug Discovery. Aug 2005, 4 (8): 649–63. PMID 16056391. S2CID 2549851. doi:10.1038/nrd1799.
- Jorgensen WL. . Science. Mar 2004, 303 (5665): 1813–8. Bibcode:2004Sci...303.1813J. PMID 15031495. S2CID 1307935. doi:10.1126/science.1096361.
- Yuan Y, Pei J, Lai L. . Current Pharmaceutical Design. Dec 2013, 19 (12): 2326–33. PMID 23082974. doi:10.2174/1381612811319120019.Yuan Y, Pei J, Lai L (Dec 2013). "Binding site detection and druggability prediction of protein targets for structure-based drug design". Current Pharmaceutical Design. 19 (12): 2326–33. doi:10.2174/1381612811319120019. PMID 23082974 (页面存档备份,存于).
- Leis S, Schneider S, Zacharias M. . Current Medicinal Chemistry. 2010, 17 (15): 1550–62. PMID 20166931. doi:10.2174/092986710790979944.
- Warren GL, Warren SD. . RSC Drug Discovery. Royal Society of Chemistry. 2011: 440–457. ISBN 978-1849731669. doi:10.1039/9781849733410-00440.
- Böhm HJ. . Journal of Computer-Aided Molecular Design. Jun 1994, 8 (3): 243–56. Bibcode:1994JCAMD...8..243B. PMID 7964925. S2CID 2491616. doi:10.1007/BF00126743.
- Liu J, Wang R. . Journal of Chemical Information and Modeling. 23 March 2015, 55 (3): 475–482. PMID 25647463. doi:10.1021/ci500731a.
- Ajay, Murcko MA. . J. Med. Chem. 1995, 38 (26): 4953–67. PMID 8544170. doi:10.1021/jm00026a001.
- Gramatica P. . RSC Drug Discovery. Royal Society of Chemistry. 2011: 466. ISBN 978-1849731669. doi:10.1039/9781849733410-00458.
- Greer J, Erickson JW, Baldwin JJ, Varney MD. . Journal of Medicinal Chemistry. Apr 1994, 37 (8): 1035–54. PMID 8164249. doi:10.1021/jm00034a001.
- Timmerman H, Gubernator K, Böhm HJ, Mannhold R, Kubinyi H. . Weinheim: Wiley-VCH. 1998. ISBN 978-3-527-29343-8.
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. . Nature Reviews. Drug Discovery. Jul 2002, 1 (7): 493–502. PMID 12120256. S2CID 2728341. doi:10.1038/nrd839.
- Klein DF. . JAMA. Mar 2008, 299 (9): 1063–5. PMID 18319418. doi:10.1001/jama.299.9.1063.