| Langer–Giedion syndrome | |
|---|---|
| Other names | Deletion 8q24.1, monosomy 8q24.1, trichorhinophalangeal syndrome type II (TRPS2), Langer–Giedion chromosome region (LGCR)[1][2] |
 | |
| A person showing the typical features of Langer–Giedion syndrome | |
| Specialty | Medical genetics |
| Differential diagnosis | Tricho-rhino-phalangeal syndrome Type 1, Fibrodysplasia Ossificans Progressiva, Trichorhinophalangeal syndrome type 3, multiple exostoses, Legg–Calvé–Perthes disease[3] |
Langer–Giedion syndrome (LGS) is a very uncommon autosomal dominant genetic disorder caused by a deletion of a small section of material on chromosome 8. It is named after the two doctors who undertook the main research into the condition in the 1960s. Diagnosis is usually made at birth or in early childhood.
Signs and symptoms
The features associated with this condition include: mild to moderate learning difficulties, short stature, unique facial features, small head and skeletal abnormalities including bony growths projecting from the surfaces of bones.[4]
Craniofacial
Individuals with Langer–Giedion syndrome may display characteristic craniofacial abnormalities. These include a long prominent philtrum, a thin upper lip, wide spaced eyes, a bulbous nasal tip, a broad nasal bridge, wide nostrils, micrognathia, retrognathia, deep set eyes and large ears. The head itself is often unusually small in comparison to that of unaffected individuals of the same age and sex. Dental abnormalities, such as supernumerary central incisors and the absence of some teeth, may occur.[5]
Muscoskeletal
 The right foot of a person with Langer–Giedion syndrome showing the characteristic features
The right foot of a person with Langer–Giedion syndrome showing the characteristic features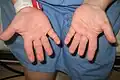 Hands of a person with Langer–Giedion syndrome showing the characteristic short fingers
Hands of a person with Langer–Giedion syndrome showing the characteristic short fingers
Langer–Giedion syndrome causes cone shaped epiphyses of the phalanges of the hands and short fingers and toes.[6] The fifth fingers are sometimes bent. Skeletal abnormalities not affecting the hands and feet may also occur. These include winged scapula, thin ribs and scoliosis. In addition, individuals with Langer–Giedion syndrome may develop hip problems similar to those seen in Legg–Calvé–Perthes disease such as progressive degeneration of the head of the thigh bone.
As affected individuals age they often develop benign boney growths called exostoses which project off the surfaces of the bones. Depending on the location of the exostoses they cause numerous complications such as compression of the spinal cord, asymmetric growth of the limbs and reduced mobility.[7]
Langer–Giedion syndrome initially causes joint hypermobility. This progresses to joint stiffness later in life when osteochondromas begin to develop, typically between infancy and mid-childhood, which decreases mobility. Hip dysplasia may be present, usually developing in early adulthood, although it can occur in infancy or childhood.[8]
Skin, hair, sweat glands and nails
Ectodermal dysplasia is a key feature of Langer–Giedion syndrome.[9]
The majority of individuals with Langer–Giedion syndrome have sparse scalp hair; this is particularly severe in males, who often experience alopecia shortly after puberty. Despite this the eyebrows may be unusually thick.[8]
Cause
The syndrome occurs when a small piece of chromosome 8's long arm, which contains a number of genes, is missing. The loss of these genes is responsible for some of the overall characteristics of Langer–Giedion syndrome.
The missing portion of the chromosome is 8q23.2–q24.1.[2] This region includes the genes TRPS1 and EXT1.
Diagnosis
Diagnosis is based on clinical findings and can be confirmed by cytogenetic testing, when the deletion is in an average of 5 Mb (millions of base pairs). Nowadays, it is a common practice to run an aCGH (array chromosome hybridization genome) study on peripheral blood of the patient, in order to delineate the extent of the loss of the genomic area, and the deleted genes.[10]
Treatment
While no genetic syndrome is capable of being cured, treatments are available for some symptoms. External fixators have been used for limbic and facial reconstructions.
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM): 150230
- 1 2 McBrien, J.; Crolla, J. A.; Huang, S.; Kelleher, J.; Gleeson, J.; Lynch, S. A. (June 2008). "Further case of microdeletion of 8q24 with phenotype overlapping Langer–Giedion withoutTRPS1 deletion". American Journal of Medical Genetics Part A. 146A (12): 1587–1592. doi:10.1002/ajmg.a.32347. PMID 18478595. S2CID 19384557.
- ↑ "Trichorhinophalangeal syndrome type ii". National Organisation for Rare Disorders. Retrieved July 7, 2021.
- ↑ Devidayal; Marwaha RK (February 2006). "Langer–Giedion Syndrome" (PDF). Indian Pediatrics. 43 (2): 174–175. PMID 16528117.
- ↑ "Trichorhinophalangeal syndrome type ii". National Organisation for Rare Disorders. Retrieved July 7, 2021.
- ↑ "trichorhinophalangeal syndrome type 2". Genetic and Rare Diseases Information Center. Retrieved July 7, 2021.
- ↑ "trichorhinophalangeal syndrome type ii". National Organisation for Rare Disorders. Retrieved July 2, 2021.
- 1 2 "trichorhinophalangeal syndrome type II". MedlinePlus. Retrieved July 7, 2021.
- ↑ Albert Schinzel; Mariluce Riegel (2013). "Long-term follow-up of four patients with Langer–Giedion syndrome". American Journal of Medical Genetics Part A. 161 (9): 2216–2225. doi:10.1002/ajmg.a.36062. PMID 23913778. S2CID 26579346.
- ↑ OMIM Entry - # 150230 - TRICHORHINOPHALANGEAL SYNDROME, TYPE II; TRPS2
External links
- Trichorhinophalangeal syndrome type 2 at NIH's Office of Rare Diseases