稳定卡宾
稳定卡宾(英语:persistent carbene或stable carbene)是一类表现出特殊稳定性的卡宾。最著名的例子和迄今最大的支类是N-杂环卡宾(简称为NHC)[1][註 1],以具有通式(R2N)2C: 的卡宾为例,其中R可以是烷基或芳基,这些基团可以相互连接得到杂环结构,并且在生物转化过程和催化领域中有独特的反应性和选择性[2],如由咪唑、咪唑啉、噻唑或三唑衍生而来的稳定卡宾。

传统的卡宾类物质被认为是反应性极强的活性中间体,只能在反应中捕获以进行研究,但随着稳定卡宾的发现出现了转机。尽管如此,稳定卡宾依然是反应性很强的物质,会自发地的变为二聚体,许多稳定卡宾在合适条件下可以作为纯物质分离,并保存一段时间。
稳定卡宾大多是单线态卡宾,其特殊稳定性可能有取代基团空间位阻大的原因[註 2]。一些单线态卡宾在热力学上是稳定的,可以分离并保存;一些则会缓慢地变为二聚物。与之相对,三线态卡宾的半衰期通常以秒为单位,可以被观测到但无法储存。
历史
早期表征
早在1957年,Ronald Breslow提出了一种相对稳定的亲核卡宾,即维生素B1(硫胺)的噻唑-2-亚基[註 3]衍生物,参与到由糠醛合成糠偶姻的维他命B1的催化循环之中[3][4]。
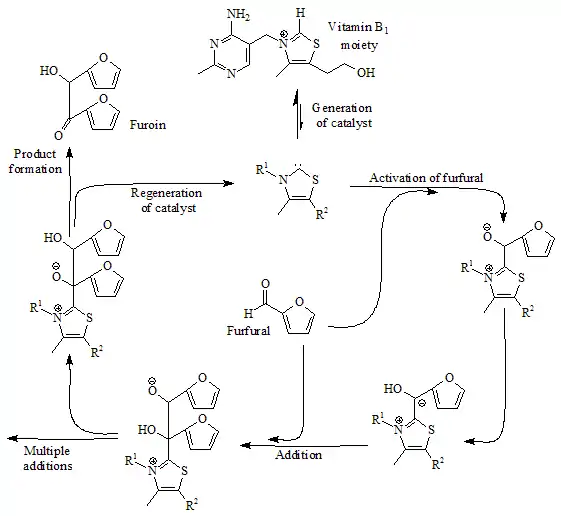
在此循环中,维生素B1通过噻唑鎓环C2上氢交换参与催化,通过重水示踪发现该C2质子在统计学平衡中快速地被氘取代。[5]
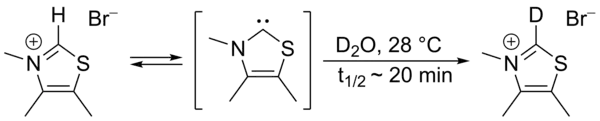
该交换过程被提出以噻唑-2-亚基的中间体进行,在2012年报告了该被称为Breslow中间体的分离 [6][7]。 到了1960年代,Hans-Werner Wanzlick及其同事推测,卡宾衍生物二氢咪唑啉-2-亚基是通过相应的2-三氯甲基-二氢咪唑真空热解脱去氯仿而得到的[8][9] [10],并提出了Wanzlick平衡,即该卡宾通过与其二聚体(一种四氨基乙烯衍生物)的化学平衡而存在。该猜想在1964年和1965年分别受到David Markham Lemal [11]和H. E. Winberg[12]的质疑。然而,后来该平衡被证明在特定情况下[13][14]可以存在。
分离
1970年,Wanzlick及其小组通过咪唑鎓盐的去质子化得到了咪唑-2-亚基卡宾[15]。Wanzlick与罗德·霍夫曼[10][16]提出由于休克尔规则的芳香性,这些咪唑基的卡宾应该会比他们的4,5-二氢物更加稳定。然而,Wanzlick并未分离出咪唑-2-亚基,只是得到了它的汞及异硫氰酸酯配合物。

1988年,Guy Bertrand等人分离出了一种磷炔,这些物质可以表示为λ3-膦基卡宾或λ5-磷乙炔[17][18]。
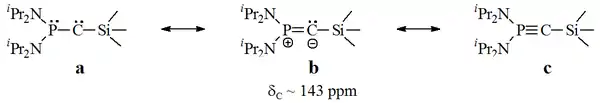
这些物质被称为推拉(push-pull)卡宾,因为磷和硅原子的电子亲和力不同,同时可以表现出卡宾和炔烃的反应性。需要说明的是,该分子的X射线结构尚未得到,并且其卡宾性质仍需更多研究。
到了1991年,Anthony Joseph Arduengo III及其同事通过咪唑鎓离子的去质子化制得了稳定的氮杂卡宾结晶[19]:

这种卡宾能在没有氧气和水的室温下稳定存在,在240–241 °C熔化但不分解,13C NMR光谱显示卡宾碳在211ppm处的信号值[20],X射线衍射显示卡宾环内的N-C键长度相比于咪唑鎓母体更长,这些键只有微弱的双键特征[21]。
至1997年制得了首个空气中稳定的氮杂卡宾,即1,3-二甲基-4,5-二氯咪唑-2-亚基[22]。 2000年,Bertrand得到了亚膦酰基型的加合卡宾,包括(亚膦酰基)(三氟甲基)卡宾,其在-30 °C溶液中稳定[23],和中等稳定性的只有一个杂原子的(氨基)(芳基)卡宾[24][25]。
影响稳定性的因素
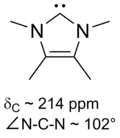
Arduengo卡宾的稳定性最初被归于N-金刚烷基较大的空间位阻阻止了二聚化,然而用甲基取代N-金刚烷基后也能得到稳定的氮杂卡宾[26]。因此,该亚基本身是热力学稳定的。咪唑鎓环上的双键也被推测赋予了体系芳香性而使整个体系变得稳定,然而这一猜想在1995年被Arduengo小组所推翻,因为他们制得了一种无双键的氮杂卡宾[27] 。关于该分子的热力学稳定性与空间位阻效应在阻止其二聚化的反应中的具体作用仍是一个争议点[28][29]。
carbene.png.webp)
自1996年第一个无环的稳定卡宾被报告[30]后,成环对其的稳定性的贡献便被认为不是必需的,通过测量此类卡宾旋转异构的势能可以判断其双键与卡宾性质,其中不受位阻影响的构象有二聚化的倾向[29][31][32]。 大多数稳定卡宾由两侧的氮中心稳定,但硫杂、氧杂的对应卡宾是例外[33][34],此类卡宾同样有二聚化的倾向。
在室温下稳定的双(二异丙基氨基)环丙烯亚基,三元卡宾环内保留了环丙烯亚基的芳香性和几何结构,这个例子证明杂原子并不是稳定卡宾的必需要素[35]。
稳定卡宾的种类
以下是迄今分离成功的稳定卡宾:
咪唑-2-亚基型
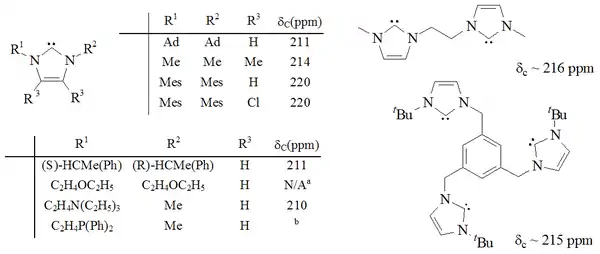
第一种被分离出来的稳定卡宾基于咪唑环结构,咪唑环2号位的碳上的氢被去除后得到,咪唑-2-亚基型的稳定卡宾是研究最多、最稳定和最为人理解的稳定卡宾。咪唑环上可以连接不同的官能团,如烷基、芳基[26]、烷氧基、烷基化氨基、烷基膦[36]、甚至是手性取代基[36]或含有两个或三个咪唑-2-亚基的分子[37][38]。
-4%252C5-dichloroimidazol-2-ylidene.png.webp)
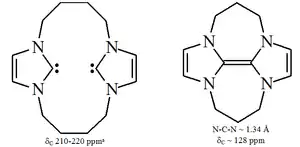
特别是用氯取代4,5位的氢后,制得了第一个有空气稳定性的卡宾[22],其独特的稳定性可能与氯取代基的吸电子效应降低了卡宾碳上孤对电子云的密度有关。咪唑环型卡宾是热力学稳定的,其中卡宾碳通常有210至230ppm的13C NMR特征化学位移值,N-C-N键角为101-102°。
三唑-5-亚基型
三唑型卡宾是另一种研究较早的稳定卡宾,三唑环拥有两种异构体,对应为1,2,3-三唑-5-亚基与1,2,4-三唑-5-亚基。

D. Enders及其同事[39]首先通过真空热解法,由2-甲氧基三唑脱去甲醇来制备1,2,4-三唑-5-亚基,该品类稳定卡宾的研究很有限,也只有少数三苯基取代物已经作为商品售卖。

三唑环基稳定卡宾是热力学稳定的,并且卡宾碳通常有210至220ppm的13C NMR特征化学位移值,通常N-C-N键角约为101°。这种卡宾的5-甲氧基三唑前体是通过甲醇钠亲核进攻处理三唑鎓制备[39],而咪唑鎓则不会与亲核试剂反应,因为会损失环上的芳香性。
其他二氨基型
上述两种稳定卡宾可以看作是二氨基型卡宾的特殊情况,二氨基型卡宾的共同点是有桥连两个氮原子的卡宾碳,许多含此类特征的稳定卡宾被Roger Alder的小组制备得到,其中许多是五或六元非芳环或双环结构[27][28][40],其中也包含非环结构的此类卡宾,或是两个独立的环[30][31][32]。

与咪唑型、三唑型的稳定卡宾不同,此种卡宾并不都是热力学稳定的,只要空间结构允许,他们就会倾向于发生二聚化[28][31]。研究[29]表明这些卡宾是通过酸催化的二聚化(如Wanzlick平衡)来完成二聚化。二氨基卡宾碳的13C NMR化学位移值介于230至270ppm间,相较于二氢咪唑-2-亚基106°的N-C-N键角,无环二氨卡宾的相应键角为121°。
一种特殊的N-杂环卡宾[41]有类似环硼氮烷的结构,构成了一个平面结构。
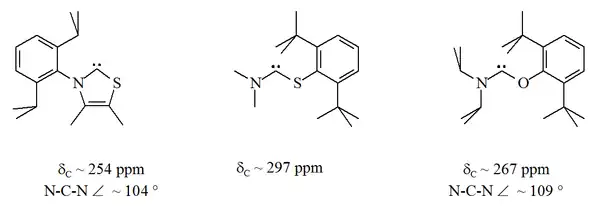
杂氨基型
二氨基型卡宾中,与卡宾碳连接的一个氮原子被其他杂原子取代后,就得到杂氨基型稳定卡宾(Heteroamino carbenes),例如氧、硫或磷[17][18][33][34]。

硫杂的类噻唑芳香杂环母体,类似于Breslow猜测的卡宾结构[42],已经被制备并经X射线衍射完成了晶体结构表征[33]。由于氧与硫是二价的,尤其是在环状结构中,卡宾碳的空间保护被限制。无环的杂氨基型稳定卡宾的13C NMR特征化学位移值在250至300ppm之间,N-C-X键角分别约为104°和109°。
非氨基型
H. D. Haztzler在1970年提出了一种由二硫化碳与缺电子乙炔衍生物反应产生瞬时的1,3-二硫杂环戊烯鎓卡宾,该卡宾随后立即二聚成四硫富瓦烯衍生物,因此,该反应的逆反应可能可以用于制备类似的卡宾[43][44]。

三线态卡宾
在2001年,Hideo Tomioka与其同事报告了一种因电子离域而相对稳定的三线态卡宾,即双(9-蒽基)卡宾,半衰期为19分钟[45][46]。

在2006年,同一小组报告了半衰期40分钟的三线态卡宾[47],此种卡宾是通过在苯中用300nm光化学分解重氮甲烷结构的前体,逸出氮气来制备的。
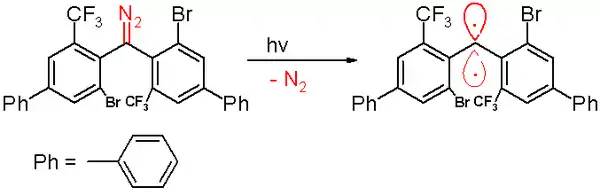
暴露于空气中会自发转化为对应的二苯酮,二苯甲烷化合物是在被环己-1,4-二烯捕获时形成,与其他稳定卡宾类似,有许多体积较大的取代基,如三氟甲基/溴代苯基,能减缓卡宾二聚化为1,1,2,2-四(苯基)烯烃的过程。根据计算机模拟,该碳原子与相邻原子的键长为138pm,键角为158.8°,苯基所在平面几乎成直角(二面角为85.7°)。
介离子卡宾
介离子卡宾与N-杂环卡宾相似,不同之处在于卡宾的典型共振式在无额外电荷的情况下无法构建,介离子卡宾也被称为异常N-杂环卡宾(aNHC)或远程N-杂环卡宾(rNHC)。部分介离子卡宾可以单独分离并在室温下稳定,但许多容易受分子间分解途径的影响。
化学性质
碱性和亲核性
咪唑-2-亚基是强碱,其共轭酸在二甲亚砜中的pKa≈24[48]:

进一步的研究表明二氨基型卡宾能使DMSO溶剂去质子化,得到的阴离子会与脒盐反应:
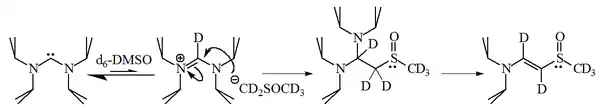
咪唑-2-亚基与1-溴己烷反应得到2-取代产物(90%)和少量对应的烯烃(10%),表明此类卡宾有相当的亲核性。在水溶液中测定的数种三唑鎓型稳定卡宾共轭酸的pKa值在16.5-17.8之间[49],酸性约强于相应的咪唑鎓型卡宾103倍[50]。
二聚化
在最初,人们认为稳定卡宾的二聚化是通过Wanzlick平衡可逆的进行,然而,咪唑-2-亚基和三唑-5-亚基能够分离并在无水和空气的情况下保存单体甚至数年之久,可能是由于此类卡宾在二聚化后会失去原本的芳香性所致。 Chen与Taton[51]通过将相应的二咪唑鎓盐去质子化制备了双系二咪唑-2-亚基,并且只在较短的亚甲基桥中才能通过去质子化得到单分子卡宾二聚物:
如果该二聚物以双卡宾的形式存在,卡宾碳上的孤电子对会被迫接近,据推测,此过程产生的相斥静电作用会使分子变得不稳定,并由此发生二聚化。 另一方面,杂氨基型卡宾(如R2N–C–OR或R2N–C–SR)和无芳香性卡宾(如R2N-C-NR2)的二聚化反应研究更加顺利[52],尽管其二聚化过程仍非常缓慢。通常这被推测是单线态卡宾高能垒的原因,不同于三线态卡宾的二聚化,单线态卡宾无法头对头地靠近,而是从卡宾孤电子对进攻空碳p轨的方向进行,由酸或金属催化:

二氨基卡宾并没有真正地二聚化,而是通过质子化的甲脒盐形成二聚物[29]。因此该反应可以被酸催化,与咪唑鎓型卡宾不同,该卡宾质子化的过程中没有失去芳香性。
反应活性
稳定卡宾的化学反应尚未探索完毕,D. Enders及其同事报道了一系列涉及三唑-5-亚基的反应,这些反应可以作为其他稳定的卡宾的参考[39][53][54]:

| a | 3,6-二苯基-1,2,4,5-四嗪,甲苯 | 92% | e | 2当量,PhNCO,甲苯,回流 | 92% | |
|---|---|---|---|---|---|---|
| b | RXH,室温 | 95–97% | f | CS2,甲苯或PhNCS,THF,室温 | 71–90% | |
| c | O2,S8,或Se,甲苯,回流 | 54–68% | g | 马来酰亚胺,THF,室温 | 47–84% | |
| d | R1CH=CHR2,THF,室温 | 25–68% | h | 丁炔二酸二甲酯,THF,回流 | 21% |
这类卡宾表现出亲核趋势(e和f )进行插入反应(b)、加成反应(c )、[2+1]环加成(d 、g 和h )、[4+1]环加成(a )。其中插入反应(b)可能是通过去质子化产生亲核体(−XR),它的进攻给人造成H-X插入的印象。
报道中异噻唑卡宾(2b)衍生自异噻唑鎓高氯酸盐(1)[55]的过程受到质疑[56],复现只能分离出2-亚胺-2H-噻丁(4),从而质疑该卡宾的稳定程度[57]。

卡宾配合物
咪唑-2-亚基、三唑-5-亚基(及少部分二氨基卡宾)能够与许多元素配位,包括碱金属、主族元素、过渡金属,甚至是镧系元素和锕系元素。下示的元素周期表列出了拥有已知相关配合物的元素,其中部分已经通过单晶X-射线进行晶体结构表征[40][58][59]。据信,稳定卡宾在作为金属配体方面的特性与膦相似,通过卡宾碳孤对电子作σ供体,但因为相邻的氮而无法很好的接受金属π电子,因此适合与相对缺电子的金属进行配位。D. Enders[60]和Wolfgang A. Herrmann[61][62]已经证明,这些卡宾在某些催化循环中是膦的可行替代配体,虽然这些替代品在活化金属催化剂方面的程度不如膦,但可以得到更耐用的催化剂。Hermann和Enders使用含咪唑和三唑型卡宾配体的催化剂取得了一定的成功[58][60][61][62] 。Grubbs[63]报道了在烯烃复分解催化剂RuCl2(PCy3)2CHPh中用咪唑-2-亚基替代膦配体(PCy3),并指出随着关环复分解反应(RCM)的增加获得了显著的“空气与水的稳定性”。含有两个和三个卡宾部分的分子已被制备作为潜在的双齿或三齿配体[37][38]。
| 元素周期表 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 族 → | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||
| ↓ 周期 | ||||||||||||||||||||
| 1 | 1 H |
2 He | ||||||||||||||||||
| 2 | 3 Li |
4 Be |
5 B |
6 C |
7 N |
8 O |
9 F |
10 Ne | ||||||||||||
| 3 | 11 Na |
12 Mg |
13 Al |
14 Si |
15 P |
16 S |
17 Cl |
18 Ar | ||||||||||||
| 4 | 19 K |
20 Ca |
21 Sc |
22 Ti |
23 V |
24 Cr |
25 Mn |
26 Fe |
27 Co |
28 Ni |
29 Cu |
30 Zn |
31 Ga |
32 Ge |
33 As |
34 Se |
35 Br |
36 Kr | ||
| 5 | 37 Rb |
38 Sr |
39 Y |
40 Zr |
41 Nb |
42 Mo |
43 Tc |
44 Ru |
45 Rh |
46 Pd |
47 Ag |
48 Cd |
49 In |
50 Sn |
51 Sb |
52 Te |
53 I |
54 Xe | ||
| 6 | 55 Cs |
56 Ba |
71 Lu |
72 Hf |
73 Ta |
74 W |
75 Re |
76 Os |
77 Ir |
78 Pt |
79 Au |
80 Hg |
81 Tl |
82 Pb |
83 Bi |
84 Po |
85 At |
86 Rn | ||
| 7 | 87 Fr |
88 Ra |
103 Lr |
104 Rf |
105 Db |
106 Sg |
107 Bh |
108 Hs |
109 Mt |
110 Ds |
111 Rg |
112 Cn |
113 Nh |
114 Fl |
115 Mc |
116 Lv |
117 Ts |
118 Og | ||
| 57 La |
58 Ce |
59 Pr |
60 Nd |
61 Pm |
62 Sm |
63 Eu |
64 Gd |
65 Tb |
66 Dy |
67 Ho |
68 Er |
69 Tm |
70 Yb | |||||||
| 89 Ac |
90 Th |
91 Pa |
92 U |
93 Np |
94 Pu |
95 Am |
96 Cm |
97 Bk |
98 Cf |
99 Es |
100 Fm |
101 Md |
102 No | |||||||
- 图例
- 有已知的卡宾配合物
- 没有已知的卡宾配合物
物理性质
目前分离出的稳定卡宾大多是低熔点的无色固体,这些卡宾在高真空度时会在很低的温度升华。卡宾碳在13C NMR光谱中的特征化学位移值在200至300ppm间,以1,3-二叔丁基-2H-咪唑-2-亚基为例,其在238pm处有一个特征峰。
与金属配位后,卡宾碳的13C共振通常会向高场转移,这取决于配合物碎片的路易斯酸强度。基于此,Huynh等人开发了一种通过trans-钯(II)-卡宾配合物的13C NMR分析供电子体强度的方法。使用13C标记的N-杂环卡宾还可用于研究混合卡宾-膦配体的配合物的顺反异构化[64]。
应用
N-杂环卡宾可以用作有机金属化学中的辅助配体,一种实际的应用是用于交叉偶联反应的钌-卡宾格拉布催化剂和氮杂环卡宾-钯配合物(NHC-Pd)[65][66][67]。NHC-金属配合物,特别是Ag(I)-NHC配合物有广泛的生物学应用[68]。
制备方法
N-杂环卡宾通常是强碱性的(咪唑-2-亚基共轭酸的pKa约为24[48])并迅速与氧气反应。虽然咪唑鎓盐对亲核试剂稳定,但其他非芳香性结构则不然(如甲脒盐)[69]。
去质子化
用强碱对卡宾前体鎓离子进行去质子化是一种可以制备绝大多数稳定卡宾的方法:
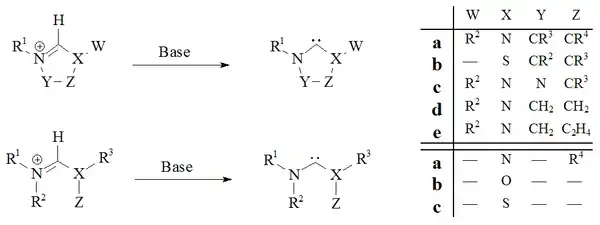
咪唑-2-亚基和二氢咪唑-2-亚基(如IMes),已经分别通过咪唑鎓盐和二氢咪唑鎓盐的去质子化制备;无环卡宾[30][31]和四氢嘧啶型卡宾[40]已经通过强均相碱去质子化制备。 反应进行的程度取决于卡宾前体的性质,这种制备方法的缺点是游离卡宾与制备过程中使用的金属离子的分离问题。
金属氢化物
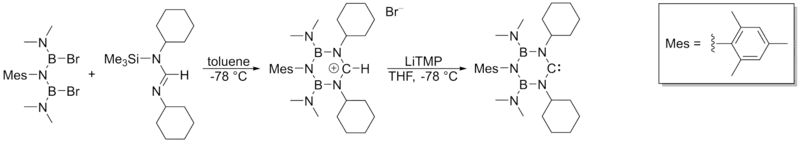
曾经氢化钠或氢化钾[27][33]被认为是理想的去质子化碱,氢化物不可逆地夺取质子,得到需要的卡宾,并能够轻松除去无机副产物和过量氢化物。在实践中,该反应通常太慢,需要加入DMSO或t-BuOH[19][26]。这些试剂通过生成叔丁醇或二甲亚砜阴离子来提高该非均相系统的反应速率。然而,这些催化剂对于制备非咪唑鎓盐的过程是无效的,氢化物中含有的氢氧根杂质也会破坏非芳香性鎓盐。 据报道[36],在−40 °C的液氨/THF混合物中使用氢化钠或氢化钾,能制得咪唑型卡宾。Arduengo及其同事[33]使用氢化钠制得了二氢咪唑型卡宾,然而,该方法尚未用于二氨基卡宾的制备。在某些方法中[26],可以加入叔丁醇钾替代金属氢化物。

氨基锂
氨基锂,如二异丙基氨基锂(LDA)和四甲基哌啶锂(LiTMP)[30][31]通常对所有类型的稳定卡宾前体有效,前提是其中没有过多的LiOH。前体鎓盐与金属六甲基二硅氨基盐(如二(三甲基硅基)氨基锂)的去质子化过程[40]也适用于大多数稳定卡宾的制备。
无金属卡宾的制备
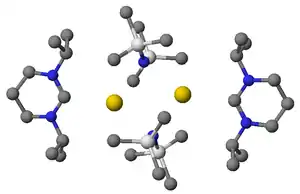
当使用过量的KHMDS作强碱去质子化甲脒盐时,会产生图示的配合物,影响产物与副产物间的分离,钾盐或钠盐副产物通常可以从溶液中沉淀分离,锂离子可以通过诸如冠醚类的穴状配体结合来去除。无金属卡宾可以通过以下方法制备:
脱硫/氧
一种制备无金属卡宾的方法是在THF中用钾对硫脲类化合物脱硫[28][70],促成该反应进行的一个原因是副产物硫化钾不溶于THF,该方法无法用于制备二聚卡宾。使用熔融钾将硫脲脱硫制备咪唑-2-亚基或二氨基卡宾的方法尚不能广泛应用,该方法可以用于制备二氢咪唑型卡宾[28]。尿素与芴卡宾脱氧得到四甲基二氨基卡宾也是一种可行的方法[71]。

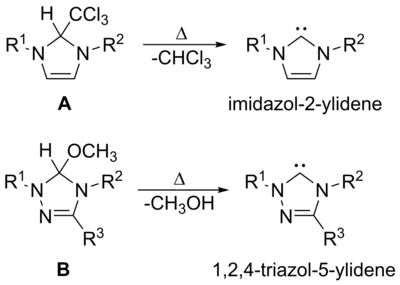
真空热解
真空热解以去除其中挥发性副产物,如甲醇或氯仿,是制备二氢咪唑型和三唑型卡宾的一种方法。历史上,Wanzlick[9]在早期尝试时就通过真空热解法从A中脱去氯仿来制备二氢咪唑-2-亚基,但这种方法也无法广泛应用。Enders小组[39]通过B的真空热解来制备三唑-5-亚基。
与双(三甲基硅烷基)汞反应
双(三甲基硅烷基)汞与氯亚胺盐和氯脒盐反应生成无金属卡宾和汞[72]:
- (CH3)3Si−Hg−Si(CH3)3 + R2N=C(Cl)−NR+
2Cl− → R2N−C−NR2 + Hg(l) + (CH3)3SiCl
光化学分解
相对稳定的三线态卡宾可以通过在苯中以300nm波长的光分解重氮甲烷前体制备。
纯化
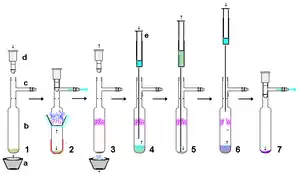
稳定卡宾反应性很强,但如果使用严格干燥、非酸性填料和无空气技术,稳定卡宾有相当的稳定性。例如,可以通过干燥的硅藻土填料过滤由氢化钾制备的稳定卡宾,来分离反应中的副产物。在小尺度上,可以使用膜式注射过滤器分离含稳定卡宾的悬浮液。稳定卡宾通常易溶于非极性溶剂,如己烷,因为没有合适的非酸性极性溶剂而难以通过重结晶方法分离。如图所示,60 °C以下、高真空升华是一种有效的提纯方法,稳定卡宾相对易于挥发,但在高温也会分解。在某些情况下,卡宾与金属离子的络合会阻碍升华。
备注
- 有时会称其为Arduengo卡宾,用以纪念第一位分离出氮杂卡宾结晶的Anthony J. Arduengo III。
- 尚有争议的内容,因为受空间效应小的NHC已经实际被制得,对稳定性判断的边界也很模糊。
- -ylidene,旧称叉基
参考文献
- Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. . Nature. 2014, 510 (7506): 485–496. Bibcode:2014Natur.510..485H. PMID 24965649. S2CID 672379. doi:10.1038/nature13384.
- Guoyong Song; Yan Su; Roy A. Periana; Robert H. Crabtree; Keli Han. . Angew. Chem. Int. Ed. 2010, 49: 912–917. doi:10.1002/anie.200905691.
- Ronald Breslow. . Chem. Ind. 1957, 26: 893.
- Ronald Breslow. . J. Am. Chem. Soc. 1958, 80 (14): 3719–3726. doi:10.1021/ja01547a064.
- R. Breslow. . J. Am. Chem. Soc. 1957, 79 (7): 1762–1763. doi:10.1021/ja01564a064.
- Berkessel A.; Elfert S.; Yatham V. R.; Neudörfl J.-M.; Schlörer N. E.; Teles J. H. . Angew. Chem. Int. Ed. 2012, 51 (49): 12370–12374. PMID 23081675. doi:10.1002/anie.201205878.
- . [2021-11-26]. (原始内容存档于2012-11-03).
- Hans-Werner Wanzlick; E. Schikora. [A new way into carbene chemistry]. Angew. Chem. 1960, 72 (14): 494. Bibcode:1960AngCh..72..494W. doi:10.1002/ange.19600721409.
- H. W. Wanzlick; E. Schikora. [A nucleophilic carbene]. Chem. Ber. 1960, 94 (9): 2389–2393. doi:10.1002/cber.19610940905.
- H. W. Wanzlick. . Angew. Chem. Int. Ed. 1962, 1 (2): 75–80. doi:10.1002/anie.196200751.
- D. M. Lemal; R. A. Lovald; K. I. Kawano. . J. Am. Chem. Soc. 1964, 86 (12): 2518–2519. doi:10.1021/ja01066a044.
- H. E. Winberg; J. E. Carnahan; D. D. Coffman; M. Brown. . J. Am. Chem. Soc. 1965, 87 (9): 2055–2056. doi:10.1021/ja01087a040.
- Denk M. K.; Hatano K.; Ma M. . Tetrahedron Lett. 1999, 40 (11): 2057–2060. doi:10.1016/S0040-4039(99)00164-1.
- Böhm Volker P. W.; Herrmann Wolfgang A. . Angew. Chem. Int. Ed. 2000, 39 (22): 4036–4038. doi:10.1002/1521-3773(20001117)39:22<4036::AID-ANIE4036>3.0.CO;2-L.
- H. W. Wanzlick; H. J. Schonherr. [Cemistry of nucleophilic carbenes, XVIII. 1) 1,3,4,5-Tetraphenylimidazolium perchlorate]. Liebigs Ann. Chem. 1970, 731: 176–179. doi:10.1002/jlac.19707310121.
- R. Gleiter; R. Hoffmann. . J. Am. Chem. Soc. 1968, 90 (20): 5457–5460. doi:10.1021/ja01022a023.
- A. Igau; H. Grutzmacher; A. Baceiredo; G. Bertrand. . J. Am. Chem. Soc. 1988, 110 (19): 6463–6466. doi:10.1021/ja00227a028.
- G. Bertrand; R. Reed. . Coord. Chem. Rev. 1994, 137: 323–355. doi:10.1016/0010-8545(94)03005-B.
- Arduengo, A.J.; Harlow, R.L.; Kline, M. . J. Am. Chem. Soc. 1991, 113 (1): 361–363. doi:10.1021/ja00001a054.
- Tapu, Daniela; Dixon, David A.; Roe, Christopher. . Chem. Rev. 12 August 2009, 109 (8): 3385–3407. PMID 19281270. doi:10.1021/cr800521g.
- Arduengo, Anthony J.; Harlow, Richard L.; Kline, Michael. . J. Am. Chem. Soc. January 1991, 113 (1): 361–363. doi:10.1021/ja00001a054.
- A. J. Arduengo; F. Davidson; H. V. R. Dias; J. R. Goerlich; D. Khasnis; W. J. Marshall; T. K. Prakasha. . J. Am. Chem. Soc. 1997, 119 (52): 12742–12749. doi:10.1021/ja973241o.
- Christophe Buron; Heinz Gornitzka; Vadim Romanenko; Guy Bertrand. . Science. 2000, 288 (5467): 834–836. Bibcode:2000Sci...288..834B. PMID 10796999. doi:10.1126/science.288.5467.834.
- Solé, Stéphane; Gornitzka, Heinz; Schoeller, Wolfgang W.; Bourissou, Didier; Bertrand, Guy. . Science. 2001, 292 (5523): 1901–1903. Bibcode:2001Sci...292.1901S. PMID 11397943. doi:10.1126/science.292.5523.1901.
- Lai Chun-Liang; Guo Wen-Hsin; Lee Ming-Tsung; Hu Ching-Han. . J. Organomet. Chem. 2005, 690 (24–25): 5867–5875. doi:10.1016/j.jorganchem.2005.07.058.
- A. J. Arduengo; H. V. R. Dias; R. L. Harlow; M. Kline. . J. Am. Chem. Soc. 1992, 114 (14): 5530–5534. doi:10.1021/ja00040a007.
- J. Arduengo; J. R. Goerlich; W. J. Marshall. . J. Am. Chem. Soc. 1995, 117 (44): 11027–11028. doi:10.1021/ja00149a034.
- M. K. Denk; A. Thadani; K. Hatano; A. J. Lough. . Angew. Chem. Int. Ed. 1997, 36 (23): 2607–2609. doi:10.1002/anie.199726071.
- Alder, RW; Chaker, L; Paolini, FP. . Chemical Communications (Cambridge, England). 2004, (19): 2172–2173. PMID 15467857. doi:10.1039/b409112d.
- R. W. Alder; P. R. Allen; M. Murray; A. G. Orpen. . Angew. Chem. Int. Ed. 1996, 35 (10): 1121–1123. doi:10.1002/anie.199611211.
- R. W. Alder; M. E. Blake. . Chem. Commun. 1997, (16): 1513–1514. doi:10.1039/a703610h.
- R. W. Alder; M. E. Blake; J. M. Oliva. . J. Phys. Chem. A. 1999, 103 (50): 11200–11211. Bibcode:1999JPCA..10311200A. doi:10.1021/jp9934228.
- A. J. Arduengo, J. R. Goerlich and W. J. Marshall. . Liebigs Ann. Chem. 1997, 1997 (2): 365–374. doi:10.1002/jlac.199719970213.
- R. W. Alder; C. P. Butts; A. G. Orpen. . J. Am. Chem. Soc. 1998, 120 (44): 11526–11527. doi:10.1021/ja9819312.
- Lavallo, Vincent; Canac, Yves; Donnadieu, Bruno; Schoeller, Wolfgang W.; Bertrand, Guy. . Science (journal). 2006, 312 (5774): 722–724. Bibcode:2006Sci...312..722L. PMC 2427275
 . PMID 16614171. doi:10.1126/science.1126675. 已忽略文本“Science ” (帮助)
. PMID 16614171. doi:10.1126/science.1126675. 已忽略文本“Science ” (帮助) - W. A. Herrmann; C. Kocher; L. J. Goossen; G. R. J. Artus. . Chem. Eur. J. 1996, 2 (12): 1627–1636. doi:10.1002/chem.19960021222.
- W. A. Herrmann; M. Elison; J. Fischer; C. Kocher; G. R. J. Artus. . Chem. Eur. J. 1996, 2 (7): 772–780. doi:10.1002/chem.19960020708.
- H. V. R. Dias; W. C. Jin. . Tetrahedron Lett. 1994, 35 (9): 1365–1366. doi:10.1016/S0040-4039(00)76219-8.
- D. Enders; K. Breuer; G. Raabe; J. Runsink; J. H. Teles; J. P. Melder; K. Ebel; S. Brode. . Angew. Chem. Int. Ed. 1995, 34 (9): 1021–1023. doi:10.1002/anie.199510211.
- R. W. Alder; M. E. Blake; C. Bortolotti; S. Buffali; C. P. Butts; E. Lineham; J. M. Oliva; A. G. Orpen; M. J. Quayle. . Chem. Commun. 1999, (3): 241–242. doi:10.1039/a808951e.
- Präsang, C; Donnadieu, B; Bertrand, G. . J. Am. Chem. Soc. 2005, 127 (29): 10182–10183. PMC 2440681 . PMID 16028925. doi:10.1021/ja052987g.
- R. Breslow. . J. Am. Chem. Soc. 1957, 79 (7): 1762–1763. doi:10.1021/ja01564a064.
- H. D. Haztzler. . J. Am. Chem. Soc. 1970, 92 (5): 1412–1413. doi:10.1021/ja00708a058.
- H. D. Hartzler. . J. Am. Chem. Soc. 1972, 95 (13): 4379–4387. doi:10.1021/ja00794a039.
- Tomioka, H; Iwamoto, E; Itakura, H; Hirai, K. . Nature. 2001, 412 (6847): 626–628. Bibcode:2001Natur.412..626T. PMID 11493917. S2CID 4373216. doi:10.1038/35088038.
- Michael Freemantle. . Chemical & Engineering News. 2001-08-13, 79 (33): 11 [2021-11-26]. doi:10.1021/cen-v079n033.p011a. (原始内容存档于2018-11-06).
- Itoh, T; Nakata, Y; Hirai, K; Tomioka, H. . J. Am. Chem. Soc. 2006, 128 (3): 957–967. PMID 16417387. doi:10.1021/ja056575j.
- R. W. Alder; P. R. Allen; S. J. Williams. . Chem. Commun. 1995, (12): 1267. doi:10.1039/c39950001267.
- Massey Richard S. (PDF). J. Am. Chem. Soc. 2012, 134 (50): 20421–20432 [2021-11-26]. PMID 23173841. doi:10.1021/ja308420c. (原始内容存档 (PDF)于2021-12-08).
- Higgins, Eleanor M.; Sherwood, Jennifer A.; Lindsay, Anita G.; Armstrong, James; Massey, Richard S.; Alder, Roger W.; O'Donoghue, Annmarie C. . Chem. Commun. 2011, 47 (5): 1559–1561. PMID 21116519. S2CID 205757477. doi:10.1039/C0CC03367G.
- T. A. Taton; P. Chen. . Angew. Chem. Int. Ed. 1996, 35 (9): 1011–1013. doi:10.1002/anie.199610111.
- Alder, Roger W.; Blake, Michael E.; Chaker, Leila; Harvey, Jeremy N.; Paolini, François; Schütz, Jan. . Angew. Chem. Int. Ed. 2004, 43 (44): 5896–5911. PMID 15457494. doi:10.1002/anie.200400654.
- Enders, D.; Breuer, K.; Runsink, J.; Teles, J.H. . Liebigs Ann. Chem. 1996, 1996 (12): 2019–2028. doi:10.1002/jlac.199619961212.
- Enders, D.; Breuer, K.; Teles, J.H.; Ebel, K. . J. Prakt. Chem. 1997, 339: 397–399. doi:10.1002/prac.19973390170.
- Wolf, J; Böhlmann, W; Findeisen, M; Gelbrich, T; Hofmann, HJ; Schulze, B. . Angew. Chem. Int. Ed. 2007, 46 (17): 3118–3121. PMID 17372997. doi:10.1002/anie.200604305.
- DeHope, A; Lavallo, V; Donnadieu, B; Schoeller, WW; Bertrand, G. . Angew. Chem. Int. Ed. 2007, 46 (36): 6922–6925. PMID 17661300. doi:10.1002/anie.200702272.
- Wolf Janine; Böhlmann Winfried; Findeisen Matthias; Gelbrich Thomas; Hofmann Hans-Jorg; Schulze Borbel. . Angew. Chem. Int. Ed. 2007, 46 (36): 6926. doi:10.1002/anie.200702746.
- Wolfgang A. Herrmann; Christian Köcher. . Angew. Chem. Int. Ed. 1997, 36 (20): 2162–2187. doi:10.1002/anie.199721621.
- Gernot Boche; Christof Hilf; Klaus Harms; Michael Marsch; John C. W. Lohrenz. . Angew. Chem. Int. Ed. 1995, 34 (4): 487–489. doi:10.1002/anie.199504871.
- D. Enders; H. Gielen; G. Raabe; J. Runsink; J. H. Teles. . Chem. Ber. 1996, 129 (12): 1483–1488. doi:10.1002/cber.19961291213.
- Wolfgang A. Herrmann; Martina Elison; Jakob Fischer; Christian Köcher; Georg R. J. Artus. . Angew. Chem. Int. Ed. 1995, 34 (21): 2371–2374. doi:10.1002/anie.199523711.
- Wolfgang A. Herrmann; Lukas J. Goossen; Christian Köcher; Georg R. J. Artus. . Angew. Chem. Int. Ed. 1996, 35 (23–24): 2805–2807. doi:10.1002/anie.199628051.
- M. Scholl; T. M. Trnka; J. P. Morgan; R. H. Grubbs. . Tetrahedron Lett. 1999, 40 (12): 2247–2250. doi:10.1016/S0040-4039(99)00217-8.
- Han Vinh Huynh; et al. . Organometallics. 2009, 28 (18): 5395–5404. doi:10.1021/om900667d .
- S. P. Nolan [editor] (2006). N-Heterocyclic carbenes in synthesis, Wiley-VCH ISBN 3-527-31400-8
- F. Glorius [editor] (2007) N-Heterocyclic carbenes in transition metal catalysis, Springer ISBN 3-540-36929-5
- Díez-González, Silvia; Marion, Nicolas; Nolan, Steven P. . Chem. Rev. 2009-08-12, 109 (8): 3612–3676. ISSN 0009-2665. PMID 19588961. S2CID 206902952. doi:10.1021/cr900074m.
- Garrison Jered C.; Youngs Wiley J. . Chem. Rev. 2005, 105 (11): 3978–4008. PMID 16277368. S2CID 43090499. doi:10.1021/cr050004s.
- Roger W. Alder; Michael E. Blake; Simone Bufali; Craig P. Butts; A. Guy Orpen; Jan Schütz; Stuart J. Williams. . J. Chem. Soc., Perkin Trans. 1. 2001, (14): 1586–1593. doi:10.1039/b104110j.
- N. Kuhn; T. Kratz. . Synthesis. 1993, 1993 (6): 561–562. doi:10.1055/s-1993-25902.
- D. Kovacs; M. S. Lee; D. Olson; J. E. Jackson. . J. Am. Chem. Soc. 1996, 118 (34): 8144–8145. doi:10.1021/ja961324j.
- Michael Otto; Salvador Conejero; Yves Canac; Vadim D. Romanenko; Valentyn Rudzevitch; Guy Bertrand. . J. Am. Chem. Soc. 2004, 126 (4): 1016–1017. PMID 14746458. doi:10.1021/ja0393325.