放射性碳定年法
放射性碳定年法[1],亦作放射性碳测年[2]、14
C测年方法[2]、放射性14
C定年法[3]等,是一种利用碳的同位素14
C的放射性来对含有有機物質的物品进行年代测定的方法。
威拉得·利比于1940年代在美国芝加哥大学发现了放射性碳定年法,并因此在1960年获得诺贝尔化学奖。这种方法基于大气中的氮与宇宙線反应不断在大气中产生放射性碳(14
C),这些14
C与大气中的氧结合形成具有放射性的二氧化碳,又通过植物的光合作用进入生物圈,然后再被动物进食摄入体内,故此所有的生物终其一生都不断地与大自然交换着14
C,直至死亡。这个交换在死后会停止,14
C的含量就会透过放射衰变逐步减少。通过测量死去动植物样本的14
C含量,例如一块木头或者一段骨头,就可以推算出动植物死亡的时间。样本越古老,可检测到的14
C含量就越少。因为14
C的半衰期(一个样本的一半衰变的时间周期)大约是5730年,所以这种方法可以可靠地测量所得到的最古老的样本大约是五万年左右,不过特殊的制备方法有时可以准确分析更古老的样本。
1960年代以来,科学家一直在研究过去的五万年里大气中的14
C比例,研究结果的数据形成了一条校准曲线,可以通过样本中放射性碳的测量值来估计样本的绝对年代。不过,不同类型的生物中14
C的含量不同(即同位素分馏),加之环境因素亦会影响生物圈中14
C的含量(即碳库效应),因此计算时必须要进行额外的校准。化石燃料(如煤和石油)的燃烧和50、60年代在近地的核试验使事情变得更复杂。因为生物体变成化石燃料所需要的时间远远比其14
C含量降低到可检测值以下所需的时间要长,因此化石燃料几乎没有14
C,结果就是自19世纪末期开始,大气中的14
C比例大幅下降。反过来,核试验大大提高了大气中14
C的含量,在1960年代中期达到了最大值,是核试验之前含量的两倍。
放射性碳的测量最早使用β粒子计数器,计算样本中14
C原子衰变时放射出的β粒子。而现在人们一般使用加速器质谱法,这个方法能统计样本中全部的14
C原子数量,而不是只统计少量在测量时发生衰变的14
C,所以这种方法可以应用在更小的样本上(如植物种子),结果出的也更快。放射性碳定年法的发展对考古学有着深远的影响——相较之前的方法,除了可以更准确地给考古遗址断代,还可以比较距离很远的地方的两个事件的发生时间。考古学史上常称之为“放射性碳革命”。地质学上,放射性碳定年法可以确定史前时代大事件的年份(如末次冰期的结束时间)。
背景
历史
1939年,劳伦斯伯克利国家实验室的马丁·卡门和塞缪尔·鲁本开始进行实验,以确定是否有机物中常见的任何元素都具有半衰期足够长的同位素,从而在生物医学研究中有价值。他们用实验室的回旋加速器合成了14
C,很快便发现这种原子的半衰期比之前设想的长的多。[4]之后,在費城富蘭克林研究所供职的塞尔日·科尔夫()预测,大气层上空的14
N与热中子反应会产生14
C。[5][6][注 1][注 2]二战期间,伯克利的威拉得·利比了解到了科尔夫的研究后,产生了可以利用放射性碳来定年的想法。[5][6]
1945年,利比来到芝加哥大学,开始着手进行放射性碳定年法相关的研究。隔年,他在一篇论文中指出,生命体中同时含有14
C和非放射性碳。[8][9]他又和其他几名研究员一道,从巴尔的摩下水道收集了些甲烷并对其进行同位素分离。分析结果表明,这批样品中含有14
C。与此相对的是,那些从石油中收集得到的甲烷因年代久远根本检测不到放射性。1947年,这些结果被发表到了《科学》上,文中阐明这意味着人们可以对含碳有机物进行年代检定。[8][10]
利比随后与詹姆斯·阿诺德一道,选了一批年代已知的样品来检验放射性碳定年理论。比如,他们从两位埃及法老左塞尔和斯尼夫鲁的坟墓中取样并测算,历史学家认为这些样本年代在公元前2625±75年,而放射性碳测得公元前2800±250年。这些结果在1949年发表到了《科学》上。[11][12][注 3]到了1960年,世界各地已建立了20多所放射性碳定年实验室。[14]该年,利比为此获得了诺贝尔化学奖。[8]
物理和化学细节
在自然界中,碳存在两种稳定且无放射性的同位素,即碳-12(12
C)和碳-13(13
C),还有一种放射性的同位素碳-14(14
C),又被称为“放射性碳”。14
C 的半衰期(给定量的14
C中的半数发生衰变所耗费的时间)大约是5,730年,因此如果没有外部的影响其在大气中的浓度会在数千年间逐渐减少,但是来自外层空间的宇宙射线会在平流层下部和对流层上部不断产生14
C,这些宇宙射线主要来自银河系,还有少部分是来自太阳。[8][15]宇宙射线会产生中子,当中子撞击氮-14原子时又产生14
C。[8]这个核反应是14
C产生的主要途径:
- n + 14
7N
→ 14
6C
+ p
其中,n表示中子,p表示質子。[16][17][注 4]14
C一旦产生就会快速与大气中的氧气结合形成一氧化碳(CO)[17],最终形成二氧化碳(CO
2)[18]:
- 14
C + O
2 → 14
CO + O
- 14
CO + OH → 14
CO
2 + H
通过这种方式产生的二氧化碳扩散至大气中,溶解在海洋里,然后通过光合作用被植物所吸收,动物再吃掉植物,放射性碳最终会分布到整个生物圈中。14
C与12
C的比例大约是1.25比1012。[19]另有约1%的碳原子是稳定同位素13
C。[8]
14
C的放射性衰变方程为:[20]
- 14
6C
→ 14
7N
+
e−
+
ν
e
通过释放一个β粒子(即电子,e−)和一个中微子(
ν
e),14
C核中的一个中子变成质子,14
C的核恢复为稳定的(非放射性)同位素14
N。[21]
原理
生物在存活期间会通过呼吸、进食等方式与其周围的自然环境不断交换碳元素,并维持一定的平衡。因此,它体内的14
C占比与周遭的大气或海洋的是一致的。生物死亡后會停止攝入14
C。然而,在原生物體內的14
C会继续衰變,導致14
C/12
C比值降低。透過这一比值和已知的14
C半衰期得知的14
C減少量(樣本越老14
C就越少),最後就可以推得此生物的死亡時間。[19]不过,受各种因素影响(见下文),测量出的14
C/12
C比值通常需要校准后才能得到准确结果。直接用14
C/12
C比值计算出的结果被称为“表观年龄”()。[22]
放射性同位素的衰变方程如下:[8]

其中N0是原始样本中同位素的原子数量(即时间t = 0处,该生物组织死亡的时刻),N是经过了时间t后样本中剩余的原子数量。[8]λ是一个常数,取决于具体的同位素;对于一个给定的同位素,λ等于该同位素原子衰变平均所需时间的期望值(即平均寿命)的倒数。[8]14
C的平均寿命是8267年,因此上面的方程也可以写作:[23]

人们假设,被测样本中的14
C/12
C原始比值应与当时大气环境中的比值相同。由于样本质量已知,人们可以通过计算样本中的原子数量来推出N0(14
C原子的数量)。随后再对当前样本中14
C的原子数量进行测算得到N,再将N0、N代入上文中的公式,即可求得样本年代t。[19]
相比于平均寿命而言,公众对于放射性元素“半衰期”(写作T1/2)的概念更为熟知,因此人们通常引入14
C的半衰期作为上文公式中的常量。[注 5]目前普遍认为14
C的半衰期是5,730±40年。[8]也就是说,约5730年后,一份样本中将会只剩下一半14
C;过了11460年后则会剩下四分之一;过了17,190年后则只剩下八分之一。
这里介绍的计算方式以多个假设为前提,只给出了理想状态下的结果。在实际应用中,14
C的含量在各个年代会有较大区别,因此以上方程计算出的结果还需要辅以其他数据进行修正。[8][25]这个过程中会用到校准曲线(见下文),会将样本中的14
C含量转化为样本的历法年龄。计算分几步进行,当中会产生一个名为“放射性碳龄”()的值。放射性碳龄是指未经校准、假设大气中14
C/12
C比值固定不变的前提下计算出来的样本年代。其数值也不用普通的历法时间来表示,而是用“放射性碳年”()为单位。[26][27]
计算放射性碳龄还要用到14
C的半衰期。利比在1949年的论文中取的是5720±47年,该值由安托瓦内特·恩格米尔等人的研究得来。[28]这一数值与现代科学界使用的半衰期值颇为相近,只是在利比发表论文后不久,该值被修改为5568±30年,[29]并沿用了十余载,又在1960年早期被改为5,730±40年,[30][31]导致大量此前发表的论文所得出的结论出错,毕竟这里所用的半衰期的误差达3%。[注 6]为了与这些早期文献保持一致,1962年英国剑桥的放射性碳大会()规定了一个新数值“利比半衰期”(),取5568年。现如今,人们在计算放射性碳龄时用的依然是利比半衰期,计算出的结果被称为“惯用年龄”()。现代使用的14
C大气含量校准曲线——IntCal,也使用了惯用年龄,因此任何使用IntCal计算出来的数值都是校正过的准确时间。在学术论文中,习惯上会将未经校准的放射性碳年用双引号括起来,以防读者混淆——一来这个值计算时用的是错误的14
C半衰期,二来该值未经大气14
C含量曲线进行校准。[26][27][33][注 7]
碳交换库

大气、生物圈、海洋中都含有碳元素,合称为“碳交换库”()。[36]各个碳交换库所贮的碳含量不同,与宇宙射线所产生的14
C充分混合的时间也不一样。这些因素会影响到各个碳库中14
C同12
C的比例,因此不同碳库中产出的样本需要被区别对待。[8]大气储有占全体碳库1.9%的碳元素。它是产生14
C的地方,这些同位素只需要花不到七年的时间就能与其他碳元素充分混合。[37]大气中14
C对12
C的比值被用作其他碳库的基准值。如果其他碳库的14
C比12
C值更低的话,要么这些碳元素年代久远导致14
C已衰变,要么这批碳元素并非来自大气。[25]比方说,海洋表面的碳元素占了全体碳库的2.4%,但其中14
C的占比只有大气的95%。[8]固然,大气和洋面之间的碳交换只需几年光景,[38]但洋面自身又与深海海水反复交融,其中后者的碳元素占了全体碳库的90%有余。[25]深海海水需要花1000年的时间才会回流至海洋表面,因此洋面的海水算是14
C含量较低的“旧”海水和14
C含量较高的“新”海水混合而来,同大气中的碳元素呈动态平衡。[25]
活跃在海洋表面的生物,其体内的14
C含量与其所居住的水层相近。由于海洋表面的14
C/12
C比值较低,海洋生物的放射性碳龄通常在400年左右。[39][40]相比之下,陆地上的生物和大气之间的碳交换更为频繁,因此其14
C/12
C的比值会与大气的保持一致。[8][注 9]这些生物的碳元素占了碳库的1.3%;全体海洋生物的总质量只有陆地生物的不到1%,因此图表中并未列出。动植物尸体形成的有机物,其质量是现有生物圈的三倍有余,由于它们不再与所处环境交换碳元素,其14
C/12
C比值比生物圈的要低。[8]
考量因素
各碳交换库的14
C/12
C比值不尽相同。如果计算过程中只考虑样本的14
C含量,那么计算结果常常不尽如人意。科学家通常会将以下额外四大类因素也纳入考量范围:
- 大气中14
C/12
C比值的变化,包括时间因素和空间因素; - 同位素分馏;
- 碳库不同部分的14
C/12
C比值变化; - 样本污染。
大气变化

在放射性碳定年法(以下简称“碳定年法”)刚刚发明的一段时间里,人们一直假设大气中14
C/12
C比值维持了几千年不变。为了验证碳定年法的准确性,人们找来了几样年代已知的文物。起初,碳定年法得到的结果还和这些文物的实际年代差不多。可后来,在测算埃及已知最古老的王朝出土文物的年代时,碳定年法给出的结果和人们预估的古埃及年代之间开始出现偏差。在当时,人们还无从判断碳定年法和预估埃及年代之间孰是孰非,但后来科学家意识到14
C/12
C比值并非此前所想的那般一成不变。在研究树木年轮后,人们对这个比值的变动有了更深刻的了解。[42][43][44]人们在测算年轮的成分、年龄后,绘制出了长达8000年的年轮数据表。[42][注 10]1960年代,汉斯·修斯利用树木年轮计算得出,埃及学家预估的年份和碳定年法给出的结论实际上是一致的。一年生植物(如玉米)的14
C/12
C比值和其生长年份的大气比值保持一致。而树木则不同,每一年都会在外表长出一圈新年轮,新的14
C并不会影响到树干里层,反而是里层的14
C会逐渐衰变。因此,每一圈年轮都会记录下其生长的那一年中大气14
C/12
C的比值。通过这种方式,科学家整理出了碳定年法所需的大气14
C/12
C比值:给定树木样本,已知树木年龄、样本中14
C的原子数量N,人们可以套用衰变公式计算出年轮生长年份的14
C原子数量N0,进而求得当年大气的14
C/12
C比值。[42][44]利用这些数据,人们得出了碳定年法所需要的各年大气14
C/12
C比值校准曲线(见下文)。[45]
19世纪,人们开始大量焚烧煤和燃油。这两种化石燃料深埋于地下、年代久远,它们的14
C基本上都消耗光了。结果,它们焚烧产生的CO
2大大降低了大气中14
C/12
C的比值。若直接照搬碳定年法对20世纪早期的物品进行测定的话,结果会比实际年代要久远得多。基于类似的原理,大城市附近的14
C浓度也会低于大气平均值。这一化石燃料效应,也被称作“修斯效应”(Suess Effect,因汉斯·修斯在1955年首次报告这一问题),对大气中14
C产生了不小的影响。如果给予充分时间让这些多出来的碳元素在各碳库中充分交换的话,14
C的活度只会减少0.2%。但由于深海海水需经多年才会与表层海水循环融合的缘故,14
C的活度实际减少了3%。[42][46]
地上核试验造成的影响更大,每次核试都会释放大量中子并产生大批14
C。从1950年开始,至1963年大气核试验被禁止这段时间内,据估算产生的14
C有好几吨。假若这批新14
C立刻均匀分布至全体碳交换库的话,大气的14
C/12
C比值只会增长寥寥几个百分点。但现实情况却是大气中的14
C含量几乎翻了一番,其中南、北半球分别在1966年、1964年达到峰值,被称作“炸弹高峰”。自那时起,碳元素逐渐交换环流至碳库的其他部分,大气14
C含量也一直在下降。[42][46][47][41]
同位素分馏
光合作用是碳元素从大气流入生物圈的主要方式。在光合作用时,12
C比13
C更好吸收,后者又比14
C更容易吸收些。这三者的吸收率不同,导致大气和植物的13
C/12
C和14
C/12
C的比值不尽相同,这一现象被称为“同位素分馏”()。[48][49]
要确定一株植物的分馏程度需要同时测量12
C和13
C两个同位素的原子量,再将所得的13
C/12
C比值与基准线“PDB”[注 11]进行比较。之所以用13
C/12
C而不是14
C/12
C,是因为前者更容易测量,而且后者的比值也很容易推导出来:13
C相对于12
C的贫化现象与它们的原子质量之差成正比,因此14
C的贫化是13
C的两倍。[25]13
C的分馏,也称作δ13C,是这样计算的:[48]
- ‰

其中“‰”代表计算结果用千分率表示。[48]由于PDB基准中的13
C含量非常高[注 12],大部分测算的δ13C值为负数。

| 材料 | δ13C通常范围 |
|---|---|
| PDB | 0‰ |
| 海洋浮游生物 | −22‰ - −17‰[49] |
| C3类植物 | −30‰ - −22‰[49] |
| C4类植物 | −15‰ - −9‰[49] |
| 大气CO 2 |
−8‰[48] |
| 海洋CO 2 |
−32‰ - −13‰[49] |
人们对海洋生物的光合作用细节了解的不够深入,目前已知它们的δ13C值取决于水温。温度较高时,CO
2不易溶于水,意味着参与光合作用的CO
2更少,分馏作用也会减少。温度高于14°C时,δ13C的值会更高。温度较低时,CO
2易溶于水,也就更容易为海洋生物获取。[49]动物的δ13C值取决于它吃什么,要是食用高δ13C的食物,其体内的δ13C也自然会更高。[48]动物自身的生物化学过程也会影响到结果。比如,骨质和骨胶原的13
C含量通常会比该生物饮食的含量高(不过二者的成因不同)。骨骼中的这一13
C富集现象也暗示了动物排泄物中的13
C比它们吃进去的13
C要少。[52]
鉴于13
C通常会占样本碳元素总量的1%,13
C/12
C的比值可通过质谱法准确量出。[25]目前,人们已通过实验测量出了许多动植物的δ13C,但在使用碳定年法测算年代时最好不要援引这些数据,而应直接测量样本的δ13C值。[48]
大气CO
2和海洋表面的碳酸盐也会有同位素分馏的现象发生,大气14
C比12
C更易溶于海洋之中。因此,海洋的14
C/12
C比值比大气的要高1.5%。这一14
C的浓缩现象与海底涌上来的“旧”海水(14
C含量低)互相抵消,导致14
C辐射的直接测量值与生物圈的其他地方大体一致。在将数据用同位素分馏校准后,人们测得海洋表面海水的表观年龄约为400年。[25][40]
海洋效应
大气的CO
2能溶于海洋表面的海水中形成碳酸根、碳酸氢根离子,从而将碳元素融进海洋中。同时,海水中的碳酸离子也会以CO
2的形式重返大气。[53]这一交换过程将大气的14
C带到海洋表面,但要让14
C分散到整个水体还需要很长一段时间。深海海水和海洋表面的海水要花很久才会混合,而且就算如此各个海域的混合程度也参差不齐。深海海水主要通过上升流回到海洋表面,这一机制在赤道地区更常见些。此外,上升流也会受到海床和海岸线形状,以及气候、风场等因素的影响。总体看来,碳元素从大气交换到洋面海水要比从洋面交换到深海中要快上许多,导致某些深海水体的海水的表观放射性碳龄能有数千年。上升流将这些“旧”海水与洋面的“新”海水混合,导致人们测得洋面海水的表观年龄为几百年(经过同位素分馏校准)。[39]海洋碳库效应并非均匀分布——平均表观年龄是400年上下,但也有地理位置相近的水域的表观年龄差距能达数百年。[39][40]这些差距可以用CALIB等软件进行人工校准。[18]海洋碳库效应也适用于各种海洋生物(如贝壳、鲸、海豹等),它们的放射性碳龄也通常有数百年。[39]
半球效应
南、北半球的大氣環流各自相对独立运作,因此两个半球的空气充分混合要花费不少时间。南半球的14
C/12
C比值比北半球的低,计算出的表观年龄也比北半球的多40年左右。[注 13]这是由于南半球的海洋面积更大,导致大气与海水之间的碳交换更为频繁。受海洋效应的影响,海水中14
C贫化得更严重些,导致南半球14
C从大气中流失的速度更快。[39][54]在南极附近,海水上升流更加强烈,也进一步加剧了南半球14
C的流失。[15]
硬水效应
如果淡水中混有年代久远碳元素(比如岩石中的碳)的话,水中的14
C/12
C比例会降低。比方说,石灰岩主要由碳酸鈣构成,流经石灰岩的河水会混入碳酸根离子。类似地,地下水也会携有其流经岩石中的碳元素。这些岩石十分古老,其14
C几乎不含任何可检测到的14
C。任何经过这里的水流的14
C/12
C比值都会下降,表观年龄甚至会拔升至数千年,并影响到生长在水中的生物的检测结果。[25]这一现象被称作“硬水效应”,因为这些水中通常含有钙离子,是硬水的特点。此外,腐殖质中的碳也能产生类似的影响,如果它们产生的时间比样本的要晚的话,腐殖质可以反过来降低淡水样本的表观年龄。[39]硬水效应的影响不一,其偏差没有统一的校准值。研究人员在面对这种情况时需要进行额外的操作来确定如何校准样本测量结果,比如分析比较淡水的沉积包覆和其相关有机物的放射性碳龄。[55]
火山
火山爆发时会喷射出大量的碳。这些碳元素原自地底,不含任何可检测的14
C,因此火山附近的14
C/12
C比值会下降。休眠火山也会喷发一些14
C含量极低的物质。那些通过光合作用吸收这些碳元素的植物,其检定的14
C/12
C比值就会更低些。比如,亚速尔富尔纳斯火山口附近的植物测得表观年龄在250年—3320年之间。[56]
样本污染
向样本中添加任何含碳物质都会对样本造成污染,导致测算结果不准确。样本中混入表观年龄低的“新”碳会导致碳定年法测算结果偏小,样本年龄越大则偏差越严重。如果一个17,000年的样本中混入了1%的“新”碳,其测算结果会减小600年;若是换成34,000年的样本,同样的样本污染会造成4000年的偏差。反之,如果样本中混入没有任何14
C残留的“老”碳,会导致碳定年法测算结果偏大,其偏差值不受样本年龄的影响。比方说,任何年代出土的样本中如果混入了1%的“老”碳,会导致其碳定年法测算结果增大80年。[57]在选定、保存、测算非常古老的样本时需要额外小心,因为样本受污染物的干扰非常大。2014年,托马斯·海厄姆等人表示,受“新碳”污染,许多文献中有关尼安德特人文物的日期过于新近。[58]
样本
用来测量年代的样本首先需要移除污染物等任何不良成分。[59]这包括移除任何可见的污染物,比如样本入土之后贯入的支根。[59]酸液、碱液可用来洗去样本上的腐植酸和碳酸污染物,但需注意不应移除含有待测碳元素的部分。[60]在此基础之上,样本需要被转化为适合测量14
C含量的形式,可以是气体、液体、固体的任何一种形式,具体取决于测量方法。[59]
取样材料
- 木质样本常常在测量之前被转化为只剩纤维素的形式,但鉴于这一步骤可能会将样本的体积缩小至原先的20%,科学家也常常对整块木材进行检定。人们还会检定木炭中的放射性碳,但也经常需要移除当中混杂的污染物。[59][60]
- 未被火烧过的骨骼样本也可用来检测,一般人们会用膠原蛋白来确定骨骼的年代,这部分蛋白在清洗掉骨头的结构材料后仍然能保存下来。在过去人们一般用羟脯胺酸,这是骨骼的构成成分氨基酸。人们一度认为羟脯胺酸很可靠,以为它只会在骨骼中出现,不过后来又在地下水中检测到了这种氨基酸。[59]对于烧过的骨骼,其检测的准确度取决于骨骼样本被烧的程度。如果骨骼只是在还原性条件下被加热的话,骨骼本身可能被碳化了,这一前提下样本通常可用碳定年法检定。[59]
- 海洋、陆生生物的外壳通常完全由碳酸钙构成,形成霰石、方解石或二者的混合物。碳酸钙极易溶解、再结晶,而再结晶生成的物质会融入其生成环境中的碳元素,这些碳元素可能来自地底。如果人们不得不检定再结晶生成的外壳的话,有时可以通过一系列测定找出外壳的原始材料。[61]此外,人们还可以检定贝壳硬蛋白,但这种蛋白只构成了外壳的1%至2%。[60]
- 泥炭的三大成分是腐植酸、腐植素和富里酸,其中腐植素不溶于碱、不易被样本环境污染,因此检定结果更为可靠。[60]泥炭检定的一大障碍是移除支根,这些根须很难从样本材料中区分出来。[59]
- 土壤中富含有机物,但很容易被新形成的腐植酸污染,因此很难用作碳定年法的样本材料。通常的做法是从土壤中筛出有机物,再用适合小样本容量的检定方法进行测算。[60]
- 其他成功用碳定年法测出年代的材料有象牙、纸、纺织品、种子颗粒与谷物、泥砖中的禾秆和陶罐中食物焦痕。[60]
准备工作与取样大小
在剔除污染物后,样本需转化成适合测量的形式。[62]如果需要气体的话,科学家通常会用CO
2。[62][63]用液体闪烁计数仪处理的样本需要转化为液态形式(通常是苯)。用加速器质谱法处理的样本,处理之前通常会转化为固体石墨靶,但有时也会用气态CO
2。[62][64]
取样大小也取决于样本类型和碳定年法具体实施技术。目前有两种技术:能测量放射性的β计数器,和加速器质谱仪。β计数器通常需要至少10克(0.35盎司)的样本。[62]加速器质谱法则更为灵敏,只需要0.5毫克的碳即可。[65]此外,年代较久的样本可考虑在检定前先用热扩散塔(thermal diffusion column)等设备富集14
C。这个制备过程需要花上一个月的时间,而且还需要将取样大小扩大到原先的十倍,但可以提高14
C/12
C比值的精度,扩大放射性碳定年法的适用年代范围。[66]
测量与结果

C
在利比首次进行放射性碳定年法实验后的数十年中,测量样本14
C含量的唯一方法是监测单个碳原子的衰变。[62]这个方法测量的是样本每单位质量每时间段中发生衰变的次数。[63]这个方法也被称作“β计数法”(),因为它测量的是衰变的14
C所放出的β粒子。[67]1970年代晚期,人们发明了一种新的计数法——加速器质谱法(,常简称),可直接测量给定样本中的14
C和12
C的原子个数。[62]AMS可以直接给出14
C/12
C比值,不需要测量样本的放射性活度(不过这两者可以互相换算)。[63]β计数法的准确度曾一度领先于AMS,但现在AMS经过不断改善已比β计数法更为准确,因此是放射性碳定年法的首选测量法。[68][69]AMS还有两大额外优势:对小样本它也能给出准确的测量值,而且它出结果的速度更快——AMS只需几分钟即可将结果控制在1%的准确度,是从前的技术远远无法比拟的。[70]
β计数法
利比用的首个粒子衰变探测仪是一台他自制的盖革计数器。他先是将样本中的碳元素转化成煤烟,然后找来了一个柱形体,将其内表面涂满了样本制得的煤烟。随后,他设法把盖革计数器的盖革管塞入这个柱形体中,尽量避免样本和线圈之间夹杂任何其他材料。[62]任何掺杂的物体都可能影响到放射强度的探测结果,因为14
C衰变产生的β粒子十分微弱,只需10微米的铝箔即可过滤掉一半β粒子。[63]
利比的方法很快被气体正比计数器取代。这种计数方法受“炸弹高峰”(见上文)的影响较小,其工作原理是探测样本中14
C产生的β粒子所制造的电离脉冲。电离脉冲与粒子能量呈正比,因而背景辐射等带来的其他电离源能够被辨识出来并直接滤掉。正比计数器会用铅或钢包裹起来,减少背景辐射、宇宙线所带来的干扰。此外,人们还会用到反符合探测仪,这些仪器安装在正比计数器外部,与计数器本身同时运作。这样一来,同时在内部和外部出现的波动就会被识别为外部干扰并过滤掉。[63]
液体闪烁计数仪是另一常用来测量14
C的仪器。它虽在1950年发明,但人们直到1960年代早期、发明了高效的苯合成方法之后才算将它真正派上用场。1970年后,这种液体计数仪成为新成立的测年实验室的宠儿。液体闪烁计数仪的工作原理是让14
C产生的β粒子与加到苯里的荧光剂发生反应,这个过程会产生闪光并被计数仪探测到。和气体计数器类似的是,液体闪烁计数仪也会在四周安装外壳并加装反符合探测仪。[71][72]
气体正比计数器和液体闪烁计数仪都是在测量给定时间内样本产生β粒子的数量。鉴于样本质量已知,二者的测量结果都可以表示成“次数每分钟每克碳”(记作cpm/g C),或者“贝克勒尔每千克碳”(记作Bq/kg C,此为国际单位制)。另外,这两种测量方法都需要测量一个空白样本的放射性活度。空白样本是指“老”碳构成的样本,这些碳元素年代久远,不应带有任何放射性。这样一来科学家就能获知背景辐射的强度,再将此值从先前样本中测得的值减去,就能获知该样本自身的14
C活度。此外,人们还会测定一个标准样本的活度作为基准线。[73][注 14]
加速器质谱法
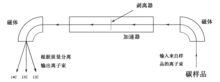
AMS仪器可清点样本中14
C和12
C的原子数量,从而直接得出14
C/12
C比值。样本通常会被制成石墨以发射C-(带一个负电荷的碳离子),并打入粒子加速器中。经过加速的碳离子会经过剥离器,失去若干电子变为阳离子(1—4个正电荷,即C+ — C4+)。随后,取决于粒子加速器的结构,这些离子会经过磁场用以弯曲其行进路径,较轻的离子比较重的离子的路径偏折得更远,因此碳离子会按其同位素种类分成一股股离子束。随后,粒子探测器会统计14
C离子束中离子的个数。不过,鉴于12
C(和用于校正的13
C)的离子束通量过大、来不及一个个探测,人们会用法拉第杯测量12
C和13
C离子束的电流大小。[76]剥离器会产生大量正电荷,能够断裂掉与14
C质量相近的分子(如13
CH)的化学键,从而排除杂质的干扰。[77]绝大多数AMS仪还能测量样本的δ13C,用来计算样本的放射性碳龄。[78]AMS相较于一般的质谱法,其优势在于可以精准区分14
N、13
CH等质量极为接近的粒子。[62]
和β计数法类似,AMS也要用到空白样本和标准样本。[76]AMS会用到两种空白样本。一种是未经过任何化学处理的“老”碳,用来检测任何仪器背景噪音;另一种被称作“处理空白样本”(),也是老碳构成的空白样本,但会用和被测样本相同的化学手法处理。背景噪音样本上出现的14
C信号通常要么来自12
CH
2和13
CH,要么就是有的离子在探测器中未按照预期的路径行进。处理空白样本上的14
C信号则能够检测在预处理样本时造成的污染。这些14
C信号在后续的计算中会用到。[79]
测得的12
C、13
C和14
C会用来计算Fm(,现代碳分数[80])。Fm是指样本14
C/12
C比例除以当代碳中14
C/12
C比例。其中,当代碳指排除化石燃料效应的前提下在1950年测算的14
C/12
C比例。[79]
计算
放射性碳定年法的计算步骤取决于之前的测量方法——β计数法测量的是样本的放射性,AMS测的是碳的三个同位素的比例。[79]二者的测量值均需在同位素分馏效应的基础上转化为同时期的木质样本的测量结果——木质样本的δ13C已知,为−25‰。[26]随后将转化后的测量值代入以下公式,即可计算得到放射性碳龄:[81]

注意这里的计算使用的是利比平均寿命8,033年。此值由利比半衰期5,568年算出,而非出自更准确的现代测量值5,730年。这个偏差会在之后的校准中纠正回来。[81]
误差和可靠度
放射性碳定年法通常只能测算5万年内诞生的样本,超过这个时间的样本中14
C极低,难以测量。一些碳定年法测得的更古老的样本用到了特殊的样本制备方法、大样本容量,并进行了长时间的测量。这些方法加在一起可以测定距今6万年甚至7.5万年的样本。[68]延长测量时长、控制样本容量大小能够提高结果的准确度——如果用β计数法测量某个样本需要250分钟才能让结果误差有68%的几率落在±80年之内,那么花上500分钟测量但是14
C的样本容量减少一半也可得到同样的置信区间。[82]
碳定年法的测定精度通常为平均值附近一个標準差(记作1σ)内。然而,1σ只有68%的置信水平,因此样本的实际真实年代可能超出测量结果给定的年代范围。1970年,大英博物馆放射性碳实验室()进行了一项实验,在6个月之内每周对同一个样本测算一次年代。测算结果服从正态分布,但彼此间差异极大,有不少测量结果的1σ年代范围互相之间不重合。比如有一个结果落在4250年—4390年,另一个落在4520年—4690年。[83]
样本制备步骤中的失误会导致结果出错。如果用作参考的现代碳样本中的苯挥发了1%,液体闪烁计数仪的结果就会比正常值少80年。[84]
校准

上文中的计算假设历史上14
C/12
C比值一成不变,计算结果以“放射性碳年”表示,而非真正的历法年份。[85]利比本人在1955年时就已经指出14
C/12
C比值很可能一直在变化,但直到后来已知历史年份和碳定年法测出来的结果分歧越来越多,人们才开始意识到测算结果需要校准才能得到真正的结果。[86]
为了确定放射性碳年所对应的历法年份,人们需要一批真实年代已知的样本,并测定这批样本的放射性碳龄。人们首先将目光投向了树木的年轮。随着每年旱季、雨季的交替,树木会生长出一圈圈薄厚不一的年轮。气候会影响局部地区的全体树木,因此科学家得以对各个树木的年轮横向比较,确定有哪些年轮的年代范围互相重合,由此进一步确定从古至今各年代对应的年轮。韦斯利·弗格森发表了首个年轮测算序列表,用的是刺果松的年轮。[44]汉斯·修斯在1967年用弗格森的数据发表了首个放射性碳定年法的校准曲线。[42][43][86]校准曲线显示了两种不同的变化,一个种是在九千年间的趋势走向,另一种是更局部的波动,通常只持续数十年。修斯说这些局部波动显示了“宇宙的动荡”(cosmic schwung),即这些波动很可能受来自地外力场的影响。不过,当时人们不清楚这些波动是真实的还是修斯杜撰的,但现在人们对宇宙线的影响已经有了更完善的认识。[42][43][87]这些局部波动现在被称作“德弗里斯效应”(),以荷兰科学家黑塞尔·德弗里斯命名。[88]
使用校准曲线时,科学家需要在纵坐标上找到实验室中测算结果给出的放射性碳龄,然后确定该纵坐标在曲线上所对应的横坐标。这一步骤与上文描述的校准曲线生成过程刚好相反。生成曲线时是给定历法年份,标出年轮等年份已知的样本的放射性碳龄;而使用曲线则是给定放射性碳龄,求出真实历法年份。[45]

在接下来的三十年里,科学家又陆陆续续发表了许多不同的校准曲线,用了多种不同的技术和统计学方法。[45]后来,人们整合出了IntCal系列校准曲线。IntCal系列在1998年首次出版,后分别在2004年、2009年、2013和2020年进行过更新,[89]使用了来自年轮、纹泥、珊瑚、植物的巨体化石、洞穴沉积物和有孔蟲的新数据。考虑到半球效应,IntCal20将北半球、南半球的数据分开罗列。南半球的校准曲线SHCAL20由独立数据汇总而来,缺少直接数据的时候会补入北半球的数据,再外加平均偏差值。此外,人们还发表了海洋校准曲线MARINE20。[33][90][91][92]
在需要对多个样本测算结果进行校准时,人们会用到贝叶斯统计方法对结果进行分析。譬如,从不同地层中提取的一系列样本,通过其他间接的考古学证据可以获知其产生的先后次序,这时人们便可以用贝叶斯分析来推测出异常值,还可以更精确地计算概率分布。[93]贝叶斯分析最开始只能在大型计算机上进行计算,但现在人们已经可以运行OxCal等程序在个人电脑上计算日期了。[94]
多个样本的时间间隔如果已知,人们会用这些时间间隔和校准曲线互相比较,以此精准匹配样本的诞生年代。这一过程也被称作“波动匹配”(),其准确度比对样本单独测算的准确度要高。[93]波动匹配在校准曲线较为平缓时比截距法(intercept method)或概率法更为有效。[95]这一技术也不一定非要用到年轮。新西兰层状火山噴發碎屑产生于人类登岛以前,科学家通过波动匹配后确定其确切年代为公元前1314±12年。[96]曲线的波动也意味着校准曲线可能给出多个年份——曲线上下摇摆会导致有两点纵坐标相同,但横坐标不同。[45]
日期格式
自首个样本的测算日期被发布以来,学界在报告样本日期时使用过多种日期格式。截至2019年,《放射性碳》期刊规定的日期格式如下:[97]
未经校准的日期应当以这个格式列出:“<实验室名>: <14
C年> ± <区间> BP”。其中:
- <实验室名>包括进行检定的实验室名称,以及样本编号
- <14
C年>是实验室测得的样本年份,单位是放射性碳年 - <区间>是实验室对放射性碳龄误差的估计,置信水平为1σ
- BP指“距今”(),此处“今”指公元1950年
举例来说,未经校准的日期“UtC-2020: 3510 ± 60 BP”表示样本在乌得勒支·范德格拉夫实验室()受检,样本编号2020,其中检定年份是距今3510年(即公元前1560年),误差±60年。此格式有时会有一定的变化,比如“10 ka BP”指距今10,000放射性碳年(即公元前8050年),人们有时还会明确列出“14
C yr BP”,以便与其他定年法(如热释光测年法)区分。[97]
经过校准的14
C年代一般会冠以“cal”以示区分。[98]《放射性碳》指定了两种校准日期格式。较为常用的格式是“cal <年代范围> <置信水平>”,其中:
- <年代范围>表示指定置信区间对应的年代
- <置信水平>表示年代范围的置信水平
比方说,“cal 1220–1281 AD (1σ)”表示此日期经过校准,结果落在公元1220年—1281年间,置信水平为一个标准差。校准日期还可以写成BP、BC的形式,此处BP依然从1950年算起。[98]用于校准日期的曲线应当使用最新的IntCal。此外,研究人员还应指出计算中使用了哪一计算机程序。[97]2014年,《放射性碳》上的一篇有关报告放射性碳日期惯例的文章建议研究人员列出对样本的处理方法,包括样本材料、预处理、质量管理方法、校准软件的版本号、校准模型、校准选项等,此外还应给出各个年代范围所对应的概率大小。[99]
应用
考古学
放射性碳定年法有助于人们进一步理解一个关键概念——文物间的考古学关联性,即考古遗址出土的多样文物之间的关系。多数情况下人们可以从文物上直接取样、鉴定,但不是所有的文物都可以进行检测。金属陪葬品就无法用碳定年法测定年代,但同一陵墓中的其他文物,像是棺椁、木炭等,一般可以假设是在同一时间入土的。这种情况下,棺椁或木炭测出来的年代就可以反映出陪葬品的年代,因为这几样物品之间存在直接功能性关系。有的样本之间的功能性关系并不强烈,但存在着其他联系,比如垃圾场里的木炭层年代与垃圾场本身的年代范围应当大体一致。[100]
树木在生长期间只有最外层的年轮会与环境之间交换碳元素,因此木质样本测定的年代取决于取样地点。这也就意味着木材样本的放射性碳龄可能比树木被砍伐的年代要久远。此外,如果木材在入土前曾多次辗转用作其他用途的话,在考古遗址中取得的木材样本所测出的年代与文物实际入土的时间差得会更远。[101]这一现象通常被称作“旧木问题”。[8]人们在英格兰维西·贝德矮林()曾发现过青铜时代的一条古道,用来铺路的木材有曾用于其他用途的痕迹。河岸发现的漂流木也可能曾经用作建材。但不是每次都能发现木材被反复使用的痕迹。其他材料也有类似的问题:比如在新石器时代,有的人会用柏油对水桶作防漏水处理。任何年代出产的柏油的放射性碳龄都远远超出实验室仪器的测定范围,因此如果研究人员不谨慎的话很容易被水桶的测定结果误导。另一个与再利用相关的问题是对材料的长时间使用,或使用完毕后很久才报废,这种情况下测出的样本年代会比实际入土年龄要大。[101]
其他领域
除了考古学外,放射性碳定年法还可用于地质学、沉积学、湖泊研究等多个领域。古植物学家和古气候学家可以通过AMS对小容量样本进行检定,从而确定沉积物序列中的花粉、少量植物材料和木炭的年代。在研究一个特定的地层时,科学家可通过地层中的有机物从而确定该地层的年代,还可以将该地层的数据拓展应用到另一地层上。[102]
放射性碳定年法还可用于测定生态系统中释放的碳元素的年代,从而可以用来监测因人类活动、气候变化从土壤中释出的碳。[103]最新的野外数据采集技术还能让科学家检定甲烷和二氧化碳的年代,二者皆为重要的温室气体。[104][105]
图克里克斯化石森林的更新世/全新世边界
更新世是一个大约始于260万年前的地质年代。当前的地质年代是全新世,始于约11,700年前、更新世结束之际。[106]20世纪的地理学家一直在确定这两个地质年代的准确界限(此处界限被划定为全球气候骤然变暖的时刻)。[106][107]美国威斯康星州图克里克斯镇有一座化石森林(现在的图克里克斯掩埋森林州立自然保护区)。研究认为森林在瓦尔德斯(Valders)冰川重新发展时被毁,这是更新世结束时该地区最后一次冰川南移。在放射性碳定年法发明之前,科学家一直在试着将图克里克斯提取的沉积物与斯堪的纳维亚的样本序列进行比对,估算出森林的年代范围在距今24,000年至19,000年之间,[106]并认定这是威斯康星冰川在彻底消退前的最后一次发展的年代,标志着更新世在北美洲的完结。[108]
1952年,利比从图克里克斯化石森林和附近其他情况类似的两地提取了样本并用放射性碳定年法测算,求得样本年代为距今11,404±350年。当时人们对放射性碳了解得不够深入,导致这一结果未经过校准。在接下来的十年里,科学家陆陆续续发表了若干结果,平均值为距今11,350年,其中较为准确的一组数据的平均值为距今11,600年。古植物学家欧内斯特·安蒂夫斯曾研究过斯堪的纳维亚纹泥,以他为代表的一众学者曾对放射性碳定年法求得的结果持异议,但最终其他地质学家决定不再采信安蒂夫斯的反对观点。1990年代,科学家将这批样本用AMS重新测了几遍,结果落在距今11,640年—11,800年间(未经校准),标准差皆为±160年。随后,从图克里克斯化石森林取得的样本被送往70多个实验室进行检定,测算结果的中位数为距今11,788±8年(2σ置信水平),校准后得到距今13,730年—13,550年。[106]图克里克斯化石森林的放射性碳定年年代让现代科学家获益匪浅,对人们了解更新世末期时北美洲冰期情况至关重要。[108]
死海古卷

1947年,科学家在死海附近的一个洞穴中发现了用希伯来语和亞拉姆語写成的死海古卷,大部分可能是犹太小众教派艾賽尼派所作。这些卷轴中包含了希伯来圣经已知的最早版本,因此对圣经文本的研究意义非凡。[109]这些卷轴被亚麻布包裹着。1955年,利比对裹着大以赛亚卷轴的亚麻布取样分析,估算年代为大约距今1,917±200年。[109][110]古文字学家根据书写风格的分析估算了21个卷轴的年代。1990年代,人们从这21个卷轴中选了一大半进行取样,再加上剩下没有经过古文字学分析的卷轴,一同送往两所AMS实验室测定。年代结果分布在公元前4世纪早期到公元4世纪中期。送检的卷轴中除了2个卷轴外,大多数卷轴测定的年代与古文字学分析的相差都不到100年。大以赛亚卷轴也在这批送检样本中,但由于对应时间段的校准曲线出现波动导致人们测算出两个年代范围:卷轴年代落在公元前355年至公元前295年的概率为15%,落在公元前210年至公元前45年的概率为84%,二者的置信水平均为2σ。但也有批评指出,在卷轴送检之前,人们用现代生产的蓖麻油对这些卷轴进行过处理以方便阅读,结果在用放射性碳定年法测算时没能将这些蓖麻油清干净,导致样本污染,测出的年代也过晚。支持和反对这一批评的论文都不少。[109]
影响
在利比1949年发表那篇《科学》论文后不久,世界各地的大学纷纷建立起了自己的放射性碳定年法实验室。在1960年时,全世界运作中的14
C研究实验室超过了20所。人们很快意识到放射性碳定年法背后的原理很经得起推敲,虽然在那时人们也有留意到碳定年法给出的结果和样本实际年代有所出入,但当时人们并不清楚造成这一现象的原因。[111]
放射性碳定年法的发明对考古学意义重大,常被称为“放射性碳革命”。[112]人类学家R·E·泰勒评价道:“14
C数据展示出的时间跨度打破了各地、各区域、各大洲的界限,为人类打开了通往史前世界的大门。”在此之前人们只能通过分析地层或石器、陶罐的类型来确定考古遗址的年代,放射性碳定年法对日期的测算能得到比这些方法更加精确的结果。此外,放射性碳定年法的结果还让科学家得以横向比对发生地点很远的两个事件,也让考古界实地数据采集方法更上一层楼(因待测样本与出土文物间的关联性变得更强)。有时候,这些实地考察方法之所以能有改善,是因为有学者希望推翻碳定年法得出的结论。放射性碳带来的另一大好处,泰勒表示,便是考古学家终于从推定文物年代的重活中解放出来,可以把精力放在那些他们真正感兴趣的研究话题上。比如,1970年代开始,考古界开始将兴趣转移到了人类行为的进化史上。[113]
放射性碳定年法还改变了人们对史前欧洲新发明传播方式的看法。此前研究人员曾认为,新发明是通过陆路传播逐渐渗透到各地的,或者是被侵略者带入到被入侵的一方。但放射性碳定年法逐渐否定了这一看法,现在人们相信这些新发明是从当地社会内部自发地出现的。这一变革也被称作“第二次放射性碳革命”。考古学家揶揄放射性碳定年法对史前英国研究的冲击,称其为给“逐渐恶化的入侵主义”的“放射……疗法”。更广泛地说,放射性碳定年法成功激起了考古学家对考古数据进行数理分析与统计的兴趣。[113]泰勒也将AMS所带来的冲击、对小样本的精准测定形容为第三次放射性碳革命的前奏。[114]
研究人员还研究过其他元素由宇宙线产生的放射性同位素,以期这些元素能在考古学中发挥作用。目前,人们研究过的同位素有3
He、10
Be、21
Ne、26
Al和36
Cl。随着AMS在1980年代的发展,人们现已能够准确测量这些同位素,并将其应用到碳定年法的实战之中,目前主要是在测量岩石的年代。[115]天然产生的放射性同位素也可以用来定年,如鉀-氬年代測定法、氩-氩年代测定法和铀-钍年代测定法等。[116]考古学家常用的年代测定技术还有热释光测年法、光释光测年法、電子自旋共振和裂变径迹定年法。另还有依靠每年生成的一层新物质来确定年代的,如树轮年代学、火山灰年代学和纹泥年代学。[117]
参见
- 考古学的定年法
注释
- 科尔夫的论文其实说的是慢中子,该词在科尔夫时代后有所变化,现指与热中子能量不同的一批中子。[7]
- 之前普遍认为14
C更可能是氘和13
C反应产生的。[4] - 2018年,人们再次检验了利比的原始样本,得出的结果与利比的结论大体一致。[13]
- 宇宙射线与地底的氮、氧相互作用也可以形成14
C,这部分同位素在某些情形下(如通过透气的地表积雪层)会流入到大气层中。不过,这一渠道产生的14
C只占了不到0.1%的总产量。[17] - 14
C的半衰期可用于推定其平均寿命(通常记作τ)。1952年时,人们认为这个值为5568±30年。[24]平均寿命和半衰期之间的关系可以用公式推出:[8] - 1950年早期还有实验测得的两个数据:~6,090年和5900±250年,但利比从未采用过这两个数据。[32]
- “放射性碳年”的定义如下:计算过程中需使用以下标准:(1)使用利比半衰期5568年,而非现如今重新测得的5730年;(2)使用國家標準技術研究所标准HOxII来定义1950年时的碳元素活度;(3)“距今XX年”的“今”从1950年算起;(4)将同位素分馏现象纳入计算的考量范围,使用一个标准的同位素比值进行计算;(5)假设14
C/12
C比值为常数,不随时间推移而改变。[34] - 碳库的百分比数据出自1990年代中叶的估算;前工业化时代的估算和这里的数据差异很大。[35]
- 海洋生物有自己的校准曲线,不然现存海洋生物经过同位素分馏校准后计算出的放射性碳龄会是400年。陆生生物也是类似的道理。
- 自那时起,年轮数据表几经修订,现已有13,900年的数据。[33]
- “PDB”指“皮迪箭石目(Pee Dee Belemnite)",美国南卡罗来纳州皮迪构造出土的化石。[50]
- PDB值是11.2372‰。[51]
- 两个最新的研究显示,过去的1000年间,南北半球的放射性碳龄差距在8—80年,平均值41±14年;过去的2000年间,这一差距为−2—83年,平均值是44±17年;再往前追溯,学者估计这个差距在50年上下。[54]
- 最常见的标准样本材料是草酸。國家標準技術研究所曾在1977年时从法国甜菜园提取了1,000英磅(450)草酸用来制作HOxII标准。[74][75]

参考资料
- 李浩. . 中国海洋大学学报(社会科学版). 2018, (3): 74–79 [2020-12-01]. (原始内容存档于2021-03-08) (中文(中国大陆)).
- 卢雪峰; 周卫建. . 地球化学. 2003, (1): 43–47 [2020-12-01]. (原始内容存档于2021-03-08) (中文(中国大陆)).
- 田婷婷; 吴中海; 张克旗; 张绪教. . 地质力学学报. 2013-09-25, 19 (3): 242–266 [2020-12-01]. (原始内容存档于2021-03-08) (中文(中国大陆)).
- Taylor & Bar-Yosef (2014), p. 268.
- Taylor & Bar-Yosef (2014), p. 269.
- . American Chemical Society. [2016-10-09]. (原始内容存档于2019-08-04).
- Korff, S.A. . Journal of the Franklin Institute. 1940, 230 (6): 777–779. Bibcode:1940TeMAE..45..133K. doi:10.1016/s0016-0032(40)90838-9.
- Bowman (1995), pp. 9–15.
- Libby, W.F. . Physical Review. 1946, 69 (11–12): 671–672. Bibcode:1946PhRv...69..671L. doi:10.1103/PhysRev.69.671.2.
- Anderson, E.C.; Libby, W.F.; Weinhouse, S.; Reid, A.F.; Kirshenbaum, A.D.; Grosse, A.V. . Science. 1947, 105 (2765): 576–577. Bibcode:1947Sci...105..576A. PMID 17746224. doi:10.1126/science.105.2735.576.
- Arnold, J.R.; Libby, W.F. . Science. 1949, 110 (2869): 678–680 [2020-06-03]. Bibcode:1949Sci...110..678A. JSTOR 1677049. PMID 15407879. doi:10.1126/science.110.2869.678. (原始内容存档于2019-01-15).
- Aitken (1990), pp. 60–61.
- Jull, A.J.T.; Pearson, C.L.; Taylor, R.E.; Southon, J.R.; Santos, G.M.; Kohl, C.P.; Hajdas, I.; Molnar, M.; Baisan, C.; Lange, T.E.; Cruz, R.; Janovics, R.; Major, I. . Radiocarbon. 2018, 60 (2): 535–548. doi:10.1017/RDC.2018.18.
- . www.c14dating.com. [2016-10-09]. (原始内容存档于2018-10-12).
- Russell, Nicola. (PDF). Glasgow, Scotland UK: University of Glasgow. 2011: 16 [2017-12-11]. (原始内容存档 (PDF)于2020-10-10).
- Bianchi & Canuel (2011), p. 35.
- Lal, D.; Jull, A.J.T. . Radiocarbon. 2001, 43 (2B): 731–742. doi:10.1017/S0033822200041394.
- Queiroz-Alves, Eduardo; Macario, Kita; Ascough, Philippa; Bronk Ramsey, Christopher. (PDF). Reviews of Geophysics. 2018, 56 (1): 278–305 [2020-06-03]. Bibcode:2018RvGeo..56..278A. doi:10.1002/2017RG000588. (原始内容存档 (PDF)于2021-02-07).
- Tsipenyuk (1997), p. 343.
- Currie, Lloyd A. . Journal of Research of the National Institute of Standards and Technology. 2004, 109 (2): 185–217. PMC 4853109
 . PMID 27366605. doi:10.6028/jres.109.013.
. PMID 27366605. doi:10.6028/jres.109.013. - Taylor & Bar-Yosef (2014), p. 33.
- Mangerud, Jan. (PDF). Boreas (奥斯陆). 1972-06-01, 1: 145 [2020-11-14] (英语).
- Aitken (1990), p. 59.
- Libby (1965), p. 42.
- Aitken (1990), pp. 61–66.
- Aitken (1990), pp. 92–95.
- Bowman (1995), p. 42.
- Engelkemeir, Antoinette G.; Hamill, W.H.; Inghram, Mark G.; Libby, W.F. . Physical Review. 1949, 75 (12): 1825. Bibcode:1949PhRv...75.1825E. doi:10.1103/PhysRev.75.1825.
- Frederick Johnson. . Memoirs of the Society for American Archaeology. 1951, (8): 1–19. JSTOR 25146610.
- H. Godwin. . Nature. 1962, 195 (4845): 984. Bibcode:1962Natur.195..984G. doi:10.1038/195984a0.
- J.van der Plicht and A.Hogg. (PDF). Quaternary Geochronology. 2006, 1 (4): 237–240 [2017-12-09]. doi:10.1016/j.quageo.2006.07.001. (原始内容存档 (PDF)于2021-02-12).
- Taylor & Bar-Yosef (2014), p. 287.
- Reimer, Paula J.; Bard, Edouard; Bayliss, Alex; Beck, J. Warren; Blackwell, Paul G.; Ramsey, Christopher Bronk; Buck, Caitlin E.; Cheng, Hai; Edwards, R. Lawrence. . Radiocarbon. 2013, 55 (4): 1869–1887. ISSN 0033-8222. doi:10.2458/azu_js_rc.55.16947.
- Taylor & Bar-Yosef (2014), pp. 26–27.
- Post (2001) pp. 128–129.
- Aitken (2003), p. 506.
- Warneck (2000), p. 690.
- Ferronsky & Polyakov (2012), p. 372.
- Bowman (1995), pp. 24–27.
- Cronin (2010), p. 35.
- Hua, Quan; Barbetti, Mike; Rakowski, Andrzej Z. . Radiocarbon. 2013, 55 (4): 2059–2072. ISSN 0033-8222. doi:10.2458/azu_js_rc.v55i2.16177.
- Bowman (1995), pp. 16–20.
- Suess (1970), p. 303.
- Taylor & Bar-Yosef (2014), pp. 50–52.
- Bowman (1995), pp. 43–49.
- Aitken (1990), pp. 71–72.
- . US Department of State. [2015-02-02]. (原始内容存档于2021-03-17).
- Bowman (1995), pp. 20–23.
- Maslin & Swann (2006), p. 246.
- Taylor & Bar-Yosef (2014), p. 125.
- Dass (2007), p. 276.
- Schoeninger (2010), p. 446.
- Libby (1965), p. 6.
- Hogg et al. (2013), p. 1898.
- Taylor & Bar-Yosef (2014), pp. 74–75.
- Pasquier-Cardina et al. (1999), pp. 200–201.
- Aitken (1990), pp. 85–86.
- Higham, T.; et al. . Nature. 2014, 512 (7514): 306–309. Bibcode:2014Natur.512..306H. PMID 25143113. doi:10.1038/nature13621.
- Bowman (1995), pp. 27–30.
- Aitken (1990), pp. 86–89.
- Šilar (2004), p. 166.
- Bowman (1995), pp. 31–37.
- Aitken (1990), pp. 76–78.
- Trumbore (1996), p. 318.
- Taylor & Bar-Yosef (2014), pp. 103–104.
- Bowman (1995), pp. 37–42.
- Walker (2005), p. 20.
- Walker (2005), p. 23.
- Killick (2014), p. 166.
- Malainey (2010), p. 96.
- Theodórsson (1996), p. 24.
- L'Annunziata & Kessler (2012), p. 424.
- Eriksson Stenström et al. (2011), p. 3.
- Terasmae (1984), p. 5.
- L'Annunziata (2007), p. 528.
- Aitken (1990), pp. 82–85.
- Wiebert (1995), p. 16.
- Tuniz, Zoppi & Barbetti (2004), p. 395.
- McNichol, A.P.; Jull, A.T.S.; Burr, G.S. . Radiocarbon. 2001, 43 (2A): 313–320 [2020-06-03]. doi:10.1017/S0033822200038169. (原始内容存档于2014-07-14).
- 钟广财; 刘頔; 李军; 张干. (PDF). 环境化学 (广州). 2015-06, 34 (6): 1027—1028 [2020-11-26]. doi:10.7524/j.issn.0254-6108.2015.06.2014111101. (原始内容存档 (PDF)于2021-02-13) (中文(中国大陆)).
- . Woods Hole Oceanographic Institution. 2007 [2013-08-27]. (原始内容存档于2020-12-20).
- Bowman (1995), pp. 38–39.
- Taylor (1987), pp. 125–126.
- Bowman (1995), pp. 40–41.
- Taylor & Bar-Yosef (2014), p. 155.
- Aitken (1990), p. 66–67.
- Taylor & Bar-Yosef (2014), p. 59.
- Taylor & Bar-Yosef (2014), pp. 53–54.
- Heaton, Timothy J.; Blaauw, Maarten; Blackwell, Paul G.; Ramsey, Christopher Bronk; Reimer, Paula J.; Scott, E. Marian. . Radiocarbon. 2020-08, 62 (4): 821–863 [2020-11-26]. ISSN 0033-8222. doi:10.1017/RDC.2020.46. (原始内容存档于2020-11-05) (英语).
- Stuiver, M.; Braziunas, T.F. . Radiocarbon. 1993, 35 (1): 137–189 [2020-06-03]. doi:10.1017/s0033822200013874. (原始内容存档于2015-02-21).
- Hogg, Alan G.; Heaton, Timothy J.; Hua, Quan; Palmer, Jonathan G.; Turney, Chris SM; Southon, John; Bayliss, Alex; Blackwell, Paul G.; Boswijk, Gretel; Ramsey, Christopher Bronk; Pearson, Charlotte. . Radiocarbon. 2020-08, 62 (4): 759–778 [2020-11-26]. ISSN 0033-8222. doi:10.1017/RDC.2020.59. (原始内容存档于2020-11-12) (英语).
- Heaton, Timothy J.; Köhler, Peter; Butzin, Martin; Bard, Edouard; Reimer, Ron W.; Austin, William E. N.; Ramsey, Christopher Bronk; Grootes, Pieter M.; Hughen, Konrad A.; Kromer, Bernd; Reimer, Paula J. . Radiocarbon. 2020-08, 62 (4): 779–820 [2020-11-26]. ISSN 0033-8222. doi:10.1017/RDC.2020.68. (原始内容存档于2020-11-03) (英语).
- Walker (2005), pp. 35–37.
- Taylor & Bar-Yosef (2014), pp. 148–149.
- Aitken1990, pp. 103–105.
- Walker (2005), pp. 207–209.
- (PDF). Radiocarbon. University of Arizona: 5–7. 2011-05-25 [2014-01-01]. (原始内容 (PDF)存档于2013-08-10).
- Taylor & Bar-Yosef (2014), p. 29.
- Millard, Andrew R. (PDF). Radiocarbon. 2014, 56 (2): 555–559 [2020-06-03]. doi:10.2458/56.17455. (原始内容存档 (PDF)于2021-02-12).
- Mook & Waterbolk (1985), pp. 48–49.
- Bowman (1995), pp. 53–54.
- Godwin, Harry. . Proceedings of the Royal Society of London B: Biological Sciences. 1961, 153 (952): 287–320. Bibcode:1961RSPSB.153..287G. doi:10.1098/rspb.1961.0001.
- Dean, Joshua F.; Garnett, Mark H.; Spyrakos, Evangelos; Billett, Michael F. . Journal of Geophysical Research: Biogeosciences. 2019, 124 (2): 328–341. Bibcode:2019JGRG..124..328D. ISSN 2169-8953. doi:10.1029/2018JG004650 (英语).
- Elder, Clayton D.; Xu, Xiaomei; Walker, Jennifer; Schnell, Jordan L.; Hinkel, Kenneth M.; Townsend-Small, Amy; Arp, Christopher D.; Pohlman, John W.; Gaglioti, Benjamin V. . Nature Climate Change. 2018, 8 (2): 166–171 [2020-11-27]. Bibcode:2018NatCC...8..166E. ISSN 1758-678X. doi:10.1038/s41558-017-0066-9. (原始内容存档于2021-03-08) (英语).
- Dean, Joshua F.; Billett, Michael F.; Murray, Callum; Garnett, Mark H. . Water Research. 2017, 115: 236–244. PMID 28284090. doi:10.1016/j.watres.2017.03.009 (英语).
- Taylor & Bar-Yosef (2014), pp. 34–37.
- Bousman & Vierra (2012), p. 4.
- Macdougall (2008), pp. 94–95.
- Taylor & Bar-Yosef (2014), pp. 38–42.
- Libby (1965), p. 84.
- Taylor & Bar-Yosef (2014), p. 288.
- Taylor (1997), p. 70.
- Taylor (1987), pp. 143–146.
- Renfrew (2014), p. 13.
- Walker (2005), pp. 77–79.
- Walker (2005), pp. 57–77.
- Walker (2005), pp. 93–162.
来源
- Aitken, M.J. . London: Longman. 1990. ISBN 978-0-582-49309-4.
- Aitken, Martin J. . Ellis, Linda (编). . New York: Garland Publishing. 2003: 505–508.
- Bianchi, Thomas S.; Canuel, Elizabeth A. . Princeton: Princeton University Press. 2011. ISBN 978-0-691-13414-7.
- Bousman, C. Britt; Vierra, Bradley J. . Bousman, C. Britt; Vierra, Bradley J. (编). . College Station, Texas: Texas A&M University Press. 2012: 1–15. ISBN 978-1-60344-760-7.
- Bowman, Sheridan.
 . London: British Museum Press. 1995 [1990]. ISBN 978-0-7141-2047-8.
. London: British Museum Press. 1995 [1990]. ISBN 978-0-7141-2047-8. - Cronin, Thomas M. . New York: Columbia University Press. 2010. ISBN 978-0-231-14494-0.
- Dass, Chhabil. . Hoboken, New Jersey: John Wiley & Sons. 2007. ISBN 978-0-471-68229-5.
- Eriksson Stenström, Kristina; Skog, Göran; Georgiadou, Elisavet; Genberg, Johan; Johansson, Anette. . Lund: Lund University. 2011 [2020-06-03]. (原始内容存档于2016-03-04).
- Ferronsky, V.I.; Polyakov, V.A. . New York: Springer. 2012. ISBN 978-94-007-2855-4.
- Killick, David. . Chapman, Robert; Alison, Wylie (编). . Abingdon, UK: Routledge. 2014: 159–172. ISBN 978-0-415-83745-3.
- L'Annunziata, Michael F. . Amsterdam: Elsevier. 2007. ISBN 978-0-444-52715-8.
- L'Annunziata, Michael F.; Kessler, Michael J. . L'Annunziata, Michael F. (编). 3rd. Oxford: Academic Press. 2012: 423–573. ISBN 978-0-12-384873-4. doi:10.1016/b978-012436603-9/50010-7.
- Libby, Willard F. 2nd (1955). Chicago: Phoenix. 1965 [1952].
- Macdougall, Doug. . Berkeley, California: University of California Press. 2008. ISBN 978-0-520-24975-2.
- Malainey, Mary E. . New York: Springer. 2010. ISBN 978-1-4419-5704-7.
- Marra, John. . Columbia University Press. 2019. ISBN 9780231186704.
- Maslin, Mark A.; Swann, George E.A. . Leng, Melanie J. (编). . Dordrecht: Springer. 2006: 227–290. ISBN 978-1-4020-2503-7. doi:10.1007/1-4020-2504-1_06.
- Mook, W.G.; Waterbolk, H.T. . Strasbourg: European Science Foundation. 1985. ISBN 978-2-903148-44-7.
- Post, Wilfred M. . Goudie, Andrew; Cuff, David J. (编). . Oxford: Oxford University Press. 2001: 127–130. ISBN 978-0-19-514518-2.
- Renfrew, Colin. . Taylor, R.E.; Bar-Yosef, Ofer (编). . Walnut Creek, California: Left Coast Press. 2014: 12–14. ISBN 978-1-59874-590-0.
- Schoeninger, Margaret J. . Larsen, Clark Spencer (编). . Oxford: Blackwell. 2010: 445–464. ISBN 978-1-4051-8900-2. doi:10.1002/9781444320039.ch25.
- Šilar, Jan. . Tykva, Richard; Berg, Dieter (编). . Dordrecht: Kluwer Academic Publishers. 2004: 150–179. ISBN 978-1-4020-1860-2.
- Suess, H.E. . Olsson, Ingrid U. (编). . New York: John Wiley & Sons. 1970: 303–311.
- Taylor, R.E. . London: Academic Press. 1987. ISBN 978-0-12-433663-6.
- Taylor, R.E. . Taylor, R.E.; Aitken, Martin J. (编). . New York: Plenum Press. 1997: 65–97. ISBN 978-0-306-45715-9.
- Taylor, R.E.; Bar-Yosef, Ofer. 2nd. Walnut Creek, California: Left Coast Press. 2014. ISBN 978-1-59874-590-0.
- Terasmae, J. . Mahaney, W.C. (编). . Amsterdam: Elsevier. 1984: 1–15. ISBN 978-0-444-42392-4.
- Theodórsson, Páll. . Singapore: World Scientific Publishing. 1996. ISBN 978-9810223151.
- Trumbore, Susan E. . Boutton, Thomas W.; Yamasaki, Shin-ichi (编). . New York: Marcel Dekker. 1996: 311–340. ISBN 978-0-8247-9699-0.
- Tsipenyuk, Yuri M. . Bristol, UK: Institute of Physics Publishing. 1997. ISBN 978-0750304221.
- Tuniz, C.; Zoppi, U.; Barbetti, M. . Martini, M.; Milazzo, M.; Piacentini, M. (编). . Amsterdam: IOS Press. 2004: 385–405. ISBN 978-1-58603-424-5.
- Walker, Mike. (PDF). Chichester: John Wiley & Sons. 2005 [2020-06-03]. ISBN 978-0-470-86927-7. (原始内容存档 (PDF)于2014-07-14).
- Warneck, Peter. . London: Academic Press. 2000. ISBN 978-0-12-735632-7.
- Wiebert, Anders. . Lund: University of Lund. 1995.