类血友病因子
类血友病因子(Von Willebrand factor,VWF),又叫血管性血友病因子,是一种参与止血,特别是血小板粘附的血液糖蛋白。VWF缺乏或缺陷会导致类血友病,并与许多其他包括血栓性血小板减少性紫癜、海德综合征和可能的溶血性尿毒综合征等疾病相关。[1]许多心血管、肿瘤、代谢(例如糖尿病)和结缔组织疾病中血浆水平升高被认为是由内皮细胞的不利变化引起的,并且可以预测血栓形成的风险增加。[2]
生化特征
结构
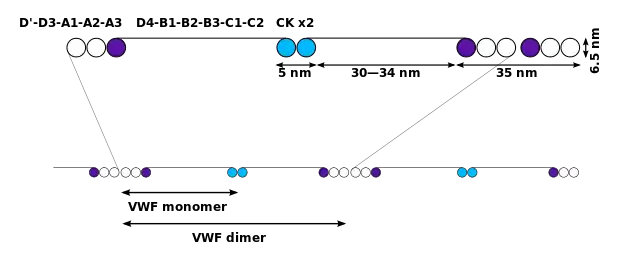
基本的VWF单体是2050个氨基酸的蛋白质。每个单体都包含许多具有特定功能的特定结构域;值得注意的要素是: [1]
- D'/D3 结构域,与因子VIII (类血友病因子D型结构域)结合。 [3]
- A1域,结合:
- A2结构域,部分展开以暴露特定ADAMTS13蛋白酶的埋藏切割位点,该蛋白酶通过产生更小的多聚体来灭活VWF。部分解折叠受到血液中剪切流、钙结合以及A2结构域C末端序列相邻“邻位二硫键”团块的影响。[4][5]
- A3结构域,与胶原蛋白结合(类血友病因子A型结构域)
- C4结构域,其中RGD基序在激活时与血小板整合素αIIbβ3结合(类血友病因子C型结构域)
- 其他C结构域,可能在ER二聚体中相互作用:较大的蛋白质在冷冻电镜显示出六个珠子(C和C样)结构域。 [3]
- “胱氨酸结”结构域(位于蛋白质的C末端),VWF与血小板衍生生长因子、转化生长因子-β和β-人绒毛膜促性腺激素共享该结构域(类血友病因子C型域)
单体被糖基化,通过半胱氨酸残基通过二硫键交联,在内质网中排列成二聚体,并在高尔基体中排列成多聚体。就糖基化而言,VWF是少数携带ABO血型系统抗原的蛋白质之一。 [1]来自高尔基体的VWF被包装成储存细胞器、内皮细胞中的Weibel-Palade 小体和血小板中的α颗粒。 [6]
VWF的多聚体可能非常大(>20,000 kDa),并且由80多个250 kDa的亚基组成。只有大的多聚体才展现出功能。VWF产生的一些裂解产物也会被分泌,但可能没有任何功能。 [1]
功能
VWF的主要功能是与其他蛋白质结合,特别是因子VIII ,并且它对于血小板粘附到伤口部位很重要。[1]由于VWF不是酶,因此不具有催化活性。
VWF与许多细胞和分子结合。一些重要结合包括: [1]
- 因子VIII在循环中不活跃时与VWF结合;当不与VWF结合时,因子VIII会迅速降解。VWF在凝血酶的作用下释放出因子VIII。在没有VWF的情况下,因子VIII的半衰期为1-2小时;当完整的VWF携带时,因子VIII的半衰期为8-12小时。
- VWF与胶原蛋白结合,例如,当胶原蛋白由于血管发生损伤而暴露在内皮细胞下方时。内皮细胞释放VWF,在血小板糖蛋白Ib/IX/V和胶原纤维之间形成额外联系。
- 当VWF与gpIX和gpV形成复合物时,它会与血小板gpIb结合;这种结合在所有情况下都会发生,但在高剪切应力(即狭窄血管中快速血流)下最有效。
- 当其他血小板受体被例如凝血酶激活,即当凝血被刺激时,VWF与其他血小板受体结合。
VWF在血液凝固中起重要作用,在狭窄血管中具有高血流剪切力的组织中最为明显,VWF在这些情况下会展开,从而减慢血小板的通过速度。因此,VWF缺乏或功能障碍(类血友病)会导致出血倾向。[1]最近的研究还表明,VWF因子参与血管新生,这可以解释为什么一些患有血管性血友病的人会出现血管畸形(主要在消化道),从而导致过度出血。 [7]
分解代谢
VWF的生物分解(分解代谢)主要由ADAMTS13酶("a disintegrin-like and metalloprotease with thrombospondin type 1 motif no. 13"的缩写 )介导。它是一种金属蛋白酶,可在A2结构域中第842位的酪氨酸和第843位(或基因的1605-1606)的蛋氨酸之间切割VWF,将多聚体分解成更小的单元,并被其他肽酶降解。 [8]
vWF在人血浆中的半衰期约为16小时;不同个体的VWF分子的糖基化变异导致这个数字可能在4.2至26小时的较大范围波动。肝细胞和巨噬细胞吸收vWF,并通过ASGPR和LRP1进行清除。SIGLEC5和CLEC4M也识别vWF。 [6]
疾病中的作用
VWF的遗传性或获得性缺陷会导致类血友病(vWD),这是一种皮肤和粘膜出血素质,导致鼻出血、月经过多和胃肠道出血。突变发生的时间点决定了出血素质的严重程度。共有三种类型(I、II、III),II型又进一步分为几个亚型。治疗取决于异常的性质和症状的严重程度。[9]大多数vWD 病例是遗传性的,但VWF异常可能是获得性的;例如,主动脉瓣狭窄与IIA型类血友病有关,导致胃肠道出血,这种关联被称为海德综合征。 [10]
在血栓性血小板减少性紫癜(TTP)和溶血性尿毒症综合征(HUS)中,ADAMTS13要么存在缺陷,要么被针对该酶的抗体抑制。这导致VWF超大多聚体的分解减少和微血管病性溶血性贫血,伴有纤维蛋白和血小板在小血管中沉积以及毛细血管坏死。在TTP中,受影响最明显的器官是大脑;在HUS中则是肾脏。[11]
在首次患有缺血性中风(由于血液凝固)的人群中,常见较高水平VWF。[12]发生率不受ADAMTS13影响,唯一重要的遗传因素是人的血型。研究发现,高血浆VWF水平是房颤患者大出血的独立预测因子。[13]
历史
VWF以芬兰医生埃里克·阿道夫·冯·维勒布兰德的名字命名,他于1926年首次描述了奥兰群岛家庭中的一种遗传性出血性疾病。尽管维勒布兰德没有确定确切的病因,但他类血友病 (vWD)与血友病和其他形式的出血素质区分开来。 [14]
20世纪50年代,vWD被证明是由血浆因子缺乏引起的(而不是由血小板疾病引起),并且在20世纪70年代,VWF蛋白被纯化。 [1] Harvey J. Weiss [15]和同事开发了一种VWF功能的定量测定方法,至今仍是VWD实验室评估的支柱。 [16]
反应
VWF已被证明与I型胶原蛋白alpha1相互作用。 [17]
最近有报道称,VWF内部的协同作用和相互作用提高了初次止血的吸附概率。这种合作可以通过计算流动的VWF一旦穿过另一吸附的VWF的吸附概率来证明。这种合作是在很宽的剪切速率范围内进行的。[18]
參考文獻
- Sadler JE. . Annual Review of Biochemistry. 1998, 67: 395–424. PMID 9759493. doi:10.1146/annurev.biochem.67.1.395
 .
. - Shahidi M. . Advances in Experimental Medicine and Biology 906. 2017: 285–306. ISBN 978-3-319-22107-6. PMID 27628010. doi:10.1007/5584_2016_122.
- Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. . Blood. July 2012, 120 (2): 449–458. PMC 3398765 . PMID 22490677. doi:10.1182/blood-2012-01-405134.
- Jakobi AJ, Mashaghi A, Tans SJ, Huizinga EG. . Nature Communications. July 2011, 2: 385. Bibcode:2011NatCo...2..385J. PMC 3144584 . PMID 21750539. doi:10.1038/ncomms1385.
- Luken BM, Winn LY, Emsley J, Lane DA, Crawley JT. . Blood. June 2010, 115 (23): 4910–4913. PMC 2890177 . PMID 20354169. doi:10.1182/blood-2009-12-257949.
- Lenting PJ, Christophe OD, Denis CV. . Blood. March 2015, 125 (13): 2019–2028. PMID 25712991. S2CID 27785232. doi:10.1182/blood-2014-06-528406 .
- Randi AM, Laffan MA. . Journal of Thrombosis and Haemostasis. January 2017, 15 (1): 13–20. PMID 27778439. S2CID 3490036. doi:10.1111/jth.13551 . hdl:10044/1/42796.
- Levy GG, Motto DG, Ginsburg D. . Blood. July 2005, 106 (1): 11–17. PMID 15774620. S2CID 25645477. doi:10.1182/blood-2004-10-4097 .
- Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. . Journal of Thrombosis and Haemostasis. October 2006, 4 (10): 2103–2114. PMID 16889557. S2CID 23875096. doi:10.1111/j.1538-7836.2006.02146.x.
- Vincentelli A, Susen S, Le Tourneau T, Six I, Fabre O, Juthier F, et al. . The New England Journal of Medicine. July 2003, 349 (4): 343–349. PMID 12878741. doi:10.1056/NEJMoa022831 .
- Moake JL. . Seminars in Hematology. January 2004, 41 (1): 4–14. PMID 14727254. doi:10.1053/j.seminhematol.2003.10.003.
- Denorme F, De Meyer SF. . Thrombosis and Haemostasis. September 2016, 116 (4): 597–604. PMID 27029413. S2CID 4964177. doi:10.1160/TH16-01-0036.
- Roldán V, Marín F, Muiña B, Torregrosa JM, Hernández-Romero D, Valdés M, et al. . Journal of the American College of Cardiology. June 2011, 57 (25): 2496–2504. PMID 21497043. doi:10.1016/j.jacc.2010.12.033 .
- von Willebrand EA. [Hereditary pseudo haemophilia]. Fin Läkaresällsk Handl. 1926, 68: 87–112 (瑞典语).
- Weiss HJ, Hoyer IW. . Science. December 1973, 182 (4117): 1149–1151. Bibcode:1973Sci...182.1149W. PMID 4127287. S2CID 41340436. doi:10.1126/science.182.4117.1149.
- Weiss HJ, Rogers J, Brand H. . The Journal of Clinical Investigation. November 1973, 52 (11): 2697–2707. PMC 302536 . PMID 4201262. doi:10.1172/JCI107464.
- Pareti FI, Fujimura Y, Dent JA, Holland LZ, Zimmerman TS, Ruggeri ZM. . The Journal of Biological Chemistry. November 1986, 261 (32): 15310–15315. PMID 3490481. doi:10.1016/S0021-9258(18)66869-3 .
- Heidari M, Mehrbod M, Ejtehadi MR, Mofrad MR. . Journal of the Royal Society, Interface. August 2015, 12 (109): 20150334. PMC 4535404 . PMID 26179989. doi:10.1098/rsif.2015.0334.
外部链接
- GeneReviews/NCBI/NIH/UW 关于冯维勒布兰德因子缺乏症的条目。包括:1 型冯维勒布兰德病、2A 型冯维勒布兰德病、2B 型冯维勒布兰德病、2M 型冯维勒布兰德病、2N 型冯维勒布兰德病、3 型冯维勒布兰德病 (页面存档备份,存于)
- PDB中UniProt可用的所有結構信息之概述:P04275 (von Willebrand factor) 在PDBe-KB。 上