| Bamford–Stevens reaction | |
|---|---|
| Named after | William Randall Bamford Thomas Stevens Stevens |
| Reaction type | Elimination reaction |
| Identifiers | |
| Organic Chemistry Portal | bamford-stevens-reaction |
| RSC ontology ID | RXNO:0000124 |
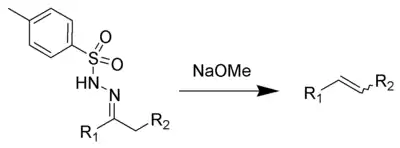
The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes.[1][2][3] It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
The treatment of tosylhydrazones with alkyl lithium reagents is called the Shapiro reaction.
Reaction mechanism
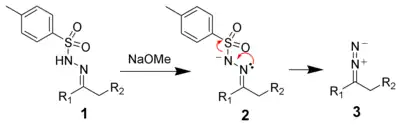
The first step of the Bamford–Stevens reaction is the formation of the diazo compound 3.[4]
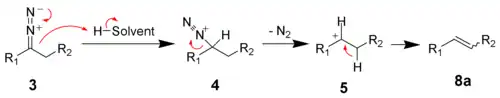
In protic solvents, the diazo compound 3 decomposes to the carbenium ion 5.
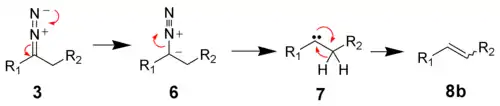
In aprotic solvents, the diazo compound 3 decomposes to the carbene 7.
Directed Bamford-Stevens reaction
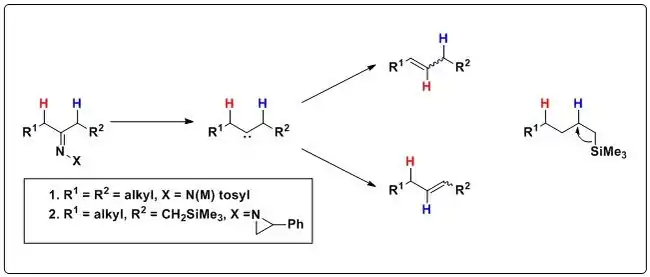
The Bamford–Stevens reaction has not proved useful for the stereoselective generation of alkenes via thermal decomposition of metallated tosylhydrazones due to the indiscriminate 1,2-rearrangement of the carbene center, which gives a mixture of products. By replacing an alkyl group with a trimethylsilyl (TMS) group on N-aziridinylimines, migration of a specific hydrogen atom can be enhanced. With the silicon atom beta to H, a σC-Si → σ*C-H stereoelectronic effect weakens the C-H bond, resulting in its exclusive migration and leading to the nearly exclusive formation of allylsilanes instead of equal amounts of allylsilanes and isomeric homoallylsilanes, analogous to the mixture of products seen in the dialkyl case, or other insertion products (i.e. cyclopropanes). See beta-silicon effect.[5][6][7]
Synthesis of 3-substituted indazoles from arynes and N-tosylhydrazones
N-tosylhydrazones can be used in a variety of synthetic procedures. Their use with arynes has been used to prepare 3-substituted indazoles via two proposed pathways. The first step is the deprotonation of the hydrazone of diazo compounds using CsF. At this point, the conjugate base could either decompose to give the diazo compound and undergo a [3+2] dipolar cycloaddition with the aryne to give the product, or a [3+2] annulation with aryne which would also give the final product. While strong bases, such as LiOtBu and Cs2CO3 are often used in this chemistry, CsF was used to facilitate the in situ generation of arynes from o-(trimethylsilyl)aryl triflates. CsF was also thought to be sufficiently basic to deprotonate the N-tosylhydrazone.[8][9]

N-tosylhydrazones as reagents for cross-coupling reactions
Barluenga and coworkers developed the first example of using N-tosylhydrazones as nucleophilic partners in cross-coupling reactions. Typically, nucleophilic reagents in coupling reactions tend to be of the organometallic variety, namely organomagnesium, -zinc, -tin, -silicon, and –boron. Combined with electrophilic aryl halides, N-tosylhydrazones can be used to prepare polysubstituted olefins under Pd-catalyzed conditions without the use of often expensive, and synthetically demanding organometallic reagents.
The scope of the reaction is wide; N-tosylhydrazones derived from aldehydes and ketones are well tolerated, which leads to both di- and trisubstituted olefins. Moreover, and variety of aryl halides are well tolerated as coupling partners including those bearing both electron-withdrawing and electron-donating groups, as well as π-rich and π-deficient aromatic heterocyclic compounds. Stereochemistry is an important element to consider when preparing polysubstituted olefins. Using hydrazones derived from linear aldehydes resulted in exclusively trans olefins, while the stereochemical outcomes of trisubstituted olefins were dependent on the size of the substituents.
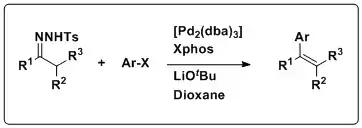
The mechanism of this transformation is thought to proceed in a manner similar to the synthesis of alkenes through the Bamford–Stevens reaction; the decomposition of N-tosylhydrazones in the presence of base to generate diazocompounds which then release nitrogen gas, yielding a carbene, which then can be quenched with an electrophile. In this case, the coupling reaction starts with the oxidative addition of the aryl halide to Pd0 catalyst to give the aryl PdII complex. The reaction of the diazocompound, generated from the hydrazone, with the PdII complex produces a Pd-carbene complex. A migratory insertion of the aryl group gives an alkyl Pd complex, which undergoes syn beta-hydride elimination to generate the trans aryl olefin and regenerate the Pd0 catalyst. This reaction has also seen utility in preparing conjugated enynes from N-tosylhydrazones and terminal alkynes under similar Pd-catalyzed reaction conditions and following the same mechanism.

Moreover, Barluenga and coworkers demonstrated a one-pot three-component coupling reaction of aldehydes or ketones, tosylhydrazides, and aryl halides in which the N-tosylhydrazone is formed in situ. This process produces stereoselective olefins in similar yields compared to the process in which preformed N-tosylhydrazones are used.[10]
Barluenga and coworkers also developed metal-free reductive coupling methodology of N-tosylhydrazones with boronic acids. The reaction tolerates a variety of functional groups on both substrates, including aromatic, heteroaromatic, aliphatic, electron-donating and electron-withdrawing substituents, and proceeds with high yields in the presence of potassium carbonate. The reaction is thought to proceed through the formation of a diazo compound that is generated from a hydrazone salt. The diazo compound could then react with the boronic acid to produce the benzylboronic acid through a boronate intermediate. An alternate pathway consists of the formation of the benzylboronic acid via a zwitterionic intermediate, followed by protodeboronation of the benzylboronic acid under basic conditions, which results in the final reductive product.

This methodology has also been extended to heteroatom nucleophiles to produce ethers and thioethers.[11][12]
A tandem rhodium-catalyzed Bamford-Stevens/thermal aliphatic Claisen rearrangement
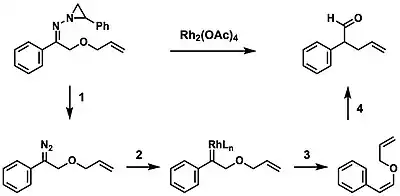
A novel process was developed by Stoltz in which the Bamford–Stevens reaction was combined with the Claisen rearrangement to produce a variety of olefin products. This transformation proceeds first by the thermal decomposition of N-aziridinylhydrazones to form the diazo compound (1), followed by a rhodium-mediated de-diazotization (2) and the syn 1,2-hydride shift (3). This substrate undergoes a thermal aliphatic Claisen rearrangement (4) to yield the product.[13][14]
Application to total synthesis
Trost et al. utilized the Bamford–Stevens reaction in their total synthesis of (–)-isoclavukerin to introduce a diene moiety found in the natural product. A bicyclic trisylhydrazone was initially subjected to Shapiro reaction conditions (alkyllithiums or LDA), which only led to uncharacterizable decomposition products. When this bicyclic trisylhydrazone was subjected to strong base (KH) and heat, however, the desired diene product was generated. Moreover, it was shown that olefin generation and the following decarboxylation could be performed in one pot. To that end, excess NaI was added, along with an elevation in temperature to facilitate the Krapcho decarboxylation.[15][16]

References
- ↑ Bamford, W. R.; Stevens, T. S. (1952). "924. The decomposition of toluene-p-sulphonylhydrazones by alkali". Journal of the Chemical Society: 4735. doi:10.1039/JR9520004735.
- ↑ Shapiro, R. H. (March 1976). "Alkenes from Tosylhydrazones". Organic Reactions. Vol. 23. New York: Wiley. pp. 405–507. ISBN 0-471-19624-X.
- ↑ Adlington, R. M.; Barrett, A. G. M. (1983). "Recent applications of the Shapiro reaction". Accounts of Chemical Research. 16 (2): 55. doi:10.1021/ar00086a004.
- ↑ Creary, X. (1986). "Tosylhydrazone salt pyrolises: phenyldiazomethanes". Organic Syntheses. 64: 207. doi:10.15227/orgsyn.064.0207. (also in the Collective Volume (1990) 7:438 (PDF))
- ↑ Sarkar, T. (1992). "Silicon-directed Bamford-Stevens reaction of β-Trimethylsilyl N-aziridinylimines". J. Chem. Soc. Chem. Commun. (17): 1184–1185. doi:10.1039/C39920001184.
- ↑ Lambert, J. (1990). "The Interaction with Silicon with Positively Charged Carbon". Tetrahedron. 46 (8): 2677–2689. doi:10.1016/s0040-4020(01)88362-9.
- ↑ Jorgensen, W. (1985). "Magnitude and origin of the .beta.-silicon effect on carbenium ions". J. Am. Chem. Soc. 107 (6): 1496–1500. doi:10.1021/ja00292a008.
- ↑ Feng, S. (2011). "Synthesis of 3-Substituted Indazoles from Arynes and N-tosylhydrazones". Org. Lett. 13 (13): 3340–3343. doi:10.1021/ol201086g. PMID 21630698.
- ↑ Pellissier, H. (2002). "The use of arynes in organic synthesis". Tetrahedron. 59 (6): 701–730. doi:10.1016/s0040-4020(02)01563-6.
- ↑ Baruenga, J. (2007). "N-tosylhydrazones as Reagents for Cross-Coupling Reactions: A Route to Polysubstituted Olefins". Angew. Chem. Int. Ed. 46 (29): 5587–5590. doi:10.1002/anie.200701815. PMID 17577897.
- ↑ Zhihui, S. (2012). "N-Tosylhydrazones: versatile reagents for metal-catalyzed and metal-free cross-coupling reactions". Chem. Soc. Rev. 41 (2): 560–572. doi:10.1039/c1cs15127d. PMID 21785803.
- ↑ Barluenga, J. (2009). "Metal-free carbon-carbon bond-forming reductive coupling between bornic acids and tosylhydrazones". Nat. Chem. 1 (6): 494–499. Bibcode:2009NatCh...1..494B. doi:10.1038/nchem.328. PMID 21378917. S2CID 35892518.
- ↑ Stoltz, B. (2002). "Non-Carbonyl-Stabilized Metallocarbenoids in Synthesis: The Development of a Tandem Rhodium-Catalyzed Bamford-Stevens/Thermal Aliphatic Claisen Rearrangement Sequence" (PDF). J. Am. Chem. Soc. 124 (42): 12426–12427. doi:10.1021/ja028020j. PMID 12381180.
- ↑ Wood, J. (1999). "Development of a Rhodium Carbenoid-Initiated Claisen Rearrangement for the Enantioselective Synthesis of α-Hydroxy Carbonyl Compounds". J. Am. Chem. Soc. 121 (8): 1748–1749. doi:10.1021/ja983294l.
- ↑ Trost, B.M. (1996). "On the Diastereoselectivity of Intramolecular Pd-Catalyzed TMM Cycloadditions. An Asymmetric Synthesis of the Perhydroazulene (–)-isoclavukerin A". J. Am. Chem. Soc. 118 (42): 10094–10105. doi:10.1021/ja961561m.
- ↑ Kurti, L.; Czako, B. (2005). Strategic Applications of Named Reactions in Organic Synthesis. El Sevier. ISBN 978-0124297852.