烯烃复分解反应
烯烃复分解反应(英語:)涉及金属催化剂存在下烯烃双键的重组,[1]自发现以来便在医药和聚合物工业中有了广泛应用。相对于其他反应,该反应副产物及废物排放少,更加环保。
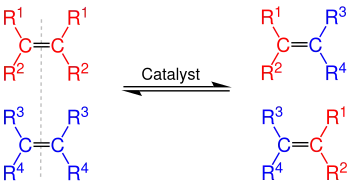
2005年的诺贝尔化学奖颁给了化学家伊夫·肖万、罗伯特·格拉布和理查德·施罗克,以表彰他们在烯烃复分解反应研究和应用方面所做出的卓越贡献。[2]
概述
烯烃复分解反应最初应用在石油工业中,以SHOP法的产物α-烯烃为原料,高温高压下生产高级烯烃。传统的反应催化剂如WCl6-EtOH-EtAlCl2,由金属卤化物与烷化剂反应制取。
烯烃复分解反应是个循环反应,过程为:首先金属卡宾配合物与烯烃反应,生成含金属杂环丁烷环系的中间体。该中间体分解,得到一个新的烯烃和新的卡宾配合物。接着后者继续发生反应,又得到原卡宾配合物。
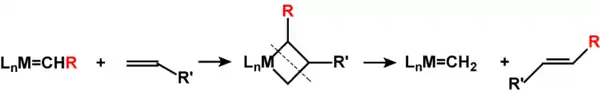
常用的催化剂都为卡宾配合物,格拉布催化剂含钌,[3]施罗克催化剂含钼或钨。[4]它们也可催化炔烃复分解反应及相关的聚合反应。
反应机理
根据伍德沃德-霍夫曼规则,两个烯烃直接发生[2+2]环加成反应是对称禁阻的,活化能很高。20世纪70年代时,Hérison和肖万提出了烯烃复分解反应的环加成机理,该机理是目前最广泛接受的反应机制。[5]其中,首先发生烯烃双键与金属卡宾配合物的[2+2]环加成反应,生成金属杂环丁烷衍生物中间体。然后该中间体经由逆环加成反应,既可得到反应物,也可得到新的烯烃和卡宾配合物。新的金属卡宾再与另一个烯烃发生类似的反应,最后生成另一个新的烯烃,并再生原金属卡宾。
金属催化剂d轨道与烯烃的相互作用降低了活化能,使烯烃复分解反应在适宜温度下就可发生,摆脱了以前多催化组分以及强路易斯酸性的反应条件。
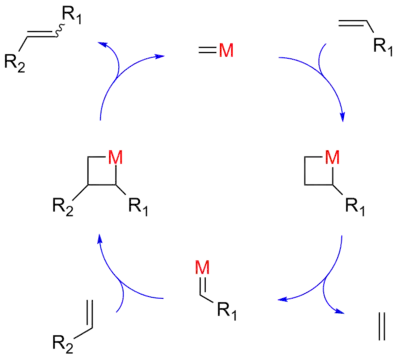
复分解反应
复分解反应又可分为以下几种重要类型:
- 交叉复分解反应
- 关环复分解反应
- 烯炔复分解反应
- 开环复分解反应
- 开环复分解聚合反应
- 非环二烯复分解反应
- 炔烃复分解反应
- 烷烃复分解反应
- 烯烃复分解反应
与大多数有机金属反应类似的是,复分解反应生成热力学控制的产物。也就是说,最终的产物比例由产物能量高低决定,符合玻尔兹曼分布。
复分解反应的驱动力往往不相同:
应用
下述反应由Hoveyda-Grubbs催化剂催化,利用了开环的交叉烯烃复分解反应:[6]
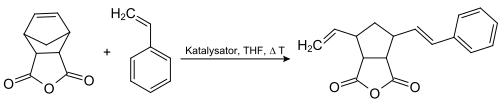
构建Epothilone分子中的大环时,采用的是烯烃复分解反应,产率很高,但双键没有选择性,生成的是E/Z等量异构体的混合物:[7]
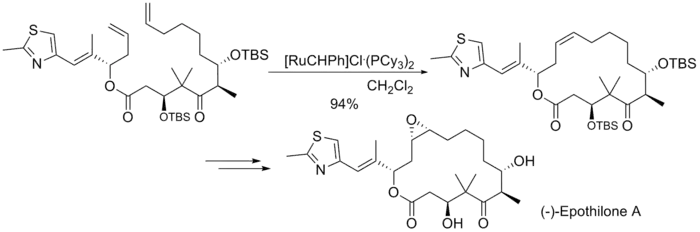 Epothilone合成
Epothilone合成
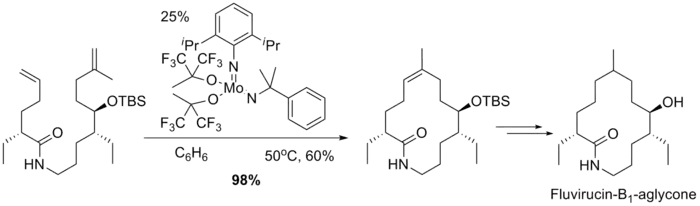
Fluvirucin-B1-Aglycone的大环合成也利用了烯烃复分解反应。但用(PCy3)2Cl2Ru=CHCH=CPh2催化时收率少于2%,只有使用钼催化剂才能以98%的产率合成关环产物,双键为Z构型:[8]
历史
烯烃复分解反应的历史可以追溯到20世纪50年代,起源于高分子化学研究。当时卡尔·齐格勒在对齐格勒-纳塔催化剂的研究中发现,一些金属催化剂存在下,乙烯“聚合”生成的产物为1-丁烯,而非应当得到的饱和碳氢长链。[10]
1960年,杜邦公司开始使用四庚基铝锂和四氯化钛为催化剂,研究降冰片烯聚合为聚降冰片烯的反应。[11]
不久后将该反应归为配位聚合反应。反应机理中,RTiX中间体先与烯烃的双键配位,生成π配合物。然后SNi协同反应使CC键断裂,生成含亚烷基-钛键的中间体,继续与第二个单体反应:

1964年,居里奥·纳塔在以钨和钼卤化物作催化剂研究环戊烯的聚合反应时,也观测到了不饱和聚合物的生成。[12]
同一年,菲利浦石油公司的研究者[13]发现,当以含有六羰基钼、六羰基钨和三氧化钼与氧化铝的混合物作催化剂时,烯烃会发生“歧化反应”,如丙烯会生成等量的乙烯和2-丁烯。他们为反应提出的机理涉及环丁烷-金属配合物的生成,具体如下:

根据两年之前诞生的伍德沃德-霍夫曼规则判断,该反应是对称性禁阻的。并且复分解反应中也从未观测到任何环丁烷衍生物的生成。因此,该机理很快便被否定了。
1967年,固特异轮胎与橡胶公司的研究人员研制出一种新颖的催化剂,由六氯化钨、乙醇和有机铝化合物EtAlMe2等组分组成,可以催化2-戊烯的复分解反应。此外,研究人员首次将该反应称为“烯烃复分解反应”。[14]
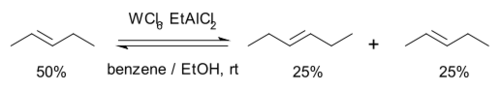
该反应中,2-戊烯很快便与2-丁烯与3-己烯形成平衡,双键并没有发生移位。而且也可以用丁烯和己烯作为原料,加入甲醇便会使反应终止。
这些研究人员还使2-丁烯与全氘代的2-丁烯反应,得到了C4H4D4,氘原子平均分配在四个碳原子上。[15] 因此,他们提出,“烯烃复分解反应”为复分解机理,而非烷基交换:
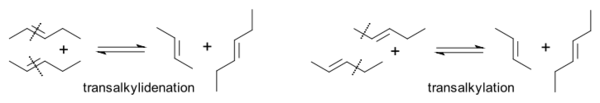
1971年,伊夫·肖万首先提出了烯烃复分解反应的金属杂四元环中间体机理,很好地解释了一些复分解反应得到统计学分布产物的现象。[16]该机理也是烯烃复分解反应目前最广泛认同的机理。
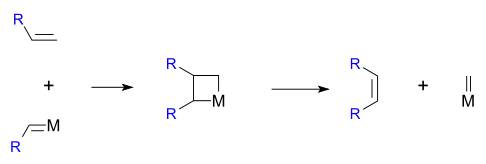
反应中涉及的金属卡宾配合物由恩斯特·奥托·菲舍尔在1964年首先制备出来。[17]
肖万机理背后的实验证据为,四氯氧化钨和四丁基锡的均相混合物催化下,环戊烯与2-戊烯的反应:
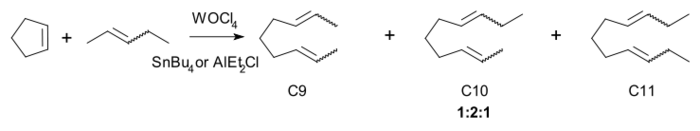
反应的三个主要产物C9、C10和C11比例为1:2:1。用更高级的低聚体反应也得到这个比例。肖万认为,卡宾由碳-金属单键的alpha-氢消除生成,例如在2-丁烯(C4)与六氯化钨和四甲基锡(C1)反应时,产物为丙烯(C3)。
同年,成功合成环丁二烯的Pettit独自提出另一套理论,[18]认为反应经由含四亚甲基的中间体,并且sp3杂化的碳原子与中心的金属原子通过多个三中心两电子键相连:
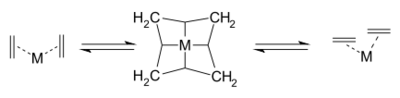
Pettit为该机理提出的实验证据是,一氧化碳可以抑制含钨金屬羰基配合物催化的4-壬烯的复分解反应。[19]
1972年起,罗伯特·格拉布开始了对复分解反应的研究。他所在的研究组使1,4-二锂代丁烷与六氯化钨反应,试图制取一个金属杂环中间体,但得到的产物与烯烃复分解反应的产物相同。因此,他也提出了金属杂环中间体的机理,只是中间体环为金属杂五元环,[20]反应为两分子反应物加到金属原子上:
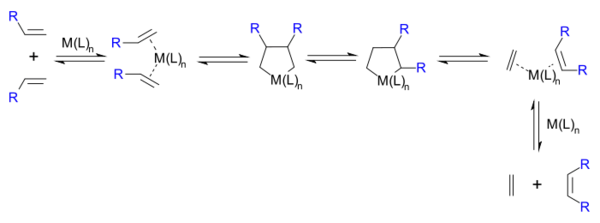
1973年,格拉布分离出二锂代丁烷与cis-双(三苯基膦)二氯合铂(II)反应生成的铂杂环中间体,进一步证实了他的机理。[21]
1975年,Katz通过让环辛烯、2-丁烯和4-辛烯的混合物与含钼的催化剂反应,观察到很快就有非对称的烃C14生成。该现象符合肖万所提出的金属杂环丁烷机理:[22]
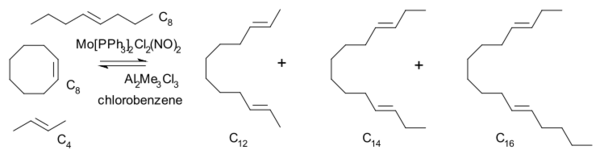
若反应的速率控制步骤为格拉布所提出的烯烃成对机理,则在主要产物C12和C16生成之后,就会立即有C12与C6进一步反应的产物产生。
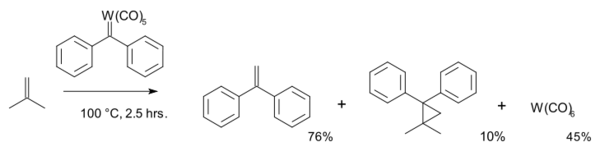
1974年,Casey首先以金属卡宾作为原料,进行烯烃复分解反应:[23]
格拉布则在1976年推翻了自己提出的机理:
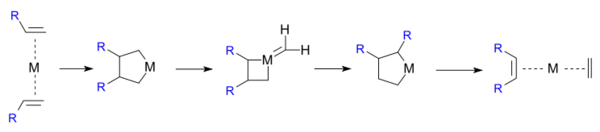
下述同位素标记的反应得到了肖万四元环中间体机理所预测的产物:[24][25]
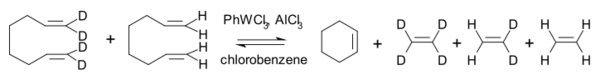
1978年开始,特伯以特伯试剂为基础,进行了很多复分解反应实验,[26]其中之一如下图所示,异丁烯和亚甲基环己烷中不同核素标记的碳原子发生交换:
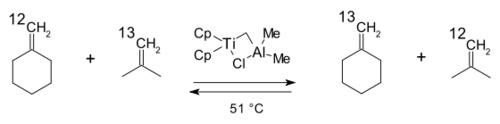
1980年,格拉布研究组利用特伯试剂与3-甲基-1-丁烯的反应,分离出第一个金属杂环丁烷衍生物:[27]
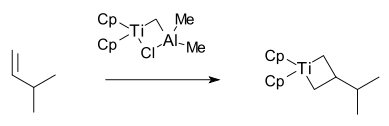

同一年,格拉布研究组证明了特伯试剂催化的降冰片烯复分解聚合反应属于活性聚合;[29]一年后,格拉布和施罗克共同发表一篇文章,描述钨卡宾配合物催化的活性聚合反应。[30]从这之后,施罗克将烯烃复分解反应的研究重点放在钨和钼催化剂上,而格拉布则着重研究含钌催化剂。他们的研究都取得了相当大的进展,相比之下,格拉布催化剂对水、酸、氧气比施罗克催化剂更加稳定。
格拉布催化剂
以Michelotti和Keaveney之前对醇类溶剂中,水合三氯化钌/锇/铱催化的降冰片烯聚合反应为基础,[31]格拉布研究组以三氯化钌、三氯化锇或钨亚烷基配合物作催化剂,成功完成了7-氧代降冰片烯衍生物的聚合反应。[32]进一步的研究表明钌(II)卡宾配合物,如(PPh3)2Cl2Ru=CHCH=CPh2具有更加有效的催化作用:[33]
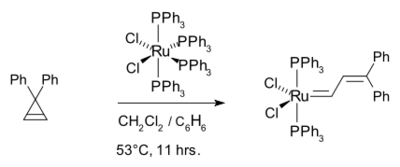
类似的还有(PCy3)2Cl2Ru=CHCH=CPh2(三苯基膦配体被三环己基膦替换),[34]也是很常用的格拉布催化剂。[35][36]
施罗克催化剂
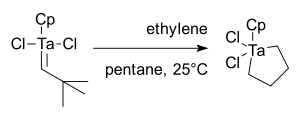
1979年,施罗克为了进一步研究钽卡宾的反应,开始涉足对烯烃复分解反应的研究。[37]最初的结果并不理想,CpTa(CHt-bu)Cl2与乙烯反应只得到一个金属杂环戊烷,并没有复分解的产物:[38]
一年后,他将原料改为PR3Ta(CHt-bu)(Ot-bu)2Cl(氯 → 叔丁氧基、环戊二烯基 → 有机膦),并用此成功催化了cis-2-戊烯的复分解反应。[39]进一步研究表明W(O)(CHt-Bu)(Cl)2(PEt)3之类的含氧钨络合物也有催化作用。[40]
1990年,Mo(NAr)(CHMe2R)(OC(CH3)(CF3)2)类型的烯烃复分解反应施罗克催化剂开始被广泛应用在有机合成中:[41][42]
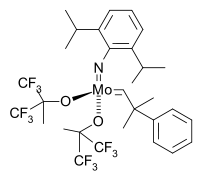
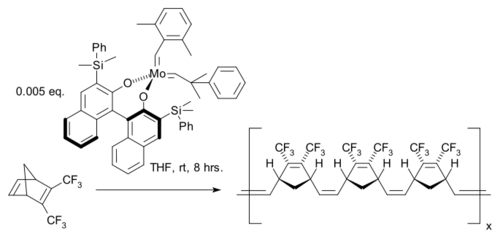
1993年结合了BINOL配体,制成了首个不对称施罗克催化剂。下面的反应中,该催化剂催化了2,5-降冰片二烯的开环复分解聚合反应,得到高度立体专一的cis同构聚合物:[43]
参考文献
- Astruc D. (abstract). New J. Chem. 2005, 29 (1): 42–56 [2008-05-23]. (原始内容存档于2008-06-23).
- (新闻稿). Nobelprize.org. 5 Oct 2005 [2008-05-23]. (原始内容存档于2018-07-10).
- Ileana Dragutan*, Valerian Dragutan*, Petru Filip. . 2005 [2008-05-23]. (原始内容存档于2006-05-12).. 105. From Arkivoc.
- R.R. Schrock. . 1986. Acc. Chem Res.
- Hérisson, J.-L.; Chauvin, Y. Macromol. Chem. 1970, 141, 161.
- A Recyclable Chiral Ru Catalyst for Enantioselective Olefin Metathesis. Efficient Catalytic Asymmetric Ring-Opening/Cross Metathesis in Air Joshua J. Van Veldhuizen, Steven B. Garber, Jason S. Kingsbury, and Amir H. Hoveyda J. Am. Chem. Soc.; 2002; 124(18) pp 4954 - 4955; (Communication) doi:10.1021/ja020259c
- H. G. Schmalz, Angew. Chem. Int. Ed. Engl. 1995, 34, 1833.
- A. F. Houri, Z. Xu, D. A. Cogan, A. H. Hoveyda, J. Am. Chem. Soc. 1995, 117, 2943; H.-G. Schmalz, Angew. Chem. Int. Ed. Engl. 1995, 34, 1833; B. M. Trost, M. A. Ceschi, B. König, Angew. Chem. Int. Ed. Engl. 1997, 36, 1486; Z. Xu, C. W. Jo-hannes, A. F. Houri, D. S. La, D. A. Cogan, G. E. Hofilena, A. H. Hoveyda, J. Am. Chem. Soc. 1997, 119, 10302.
- Ione M. Baibich, Carla Kern. . Journal of the Brazilian Chemical Society. 2002, 13 (1): 43–46 [2008-05-23]. (原始内容存档于2020-02-03).
- Polymerisation von Äthylen und anderen Olefinen Karl Ziegler, E. Holzkamp, H. Breil, H. Martin Angewandte Chemie Volume 67, Issue 16 , Pages 426 - 426 1955 doi:10.1002/ange.19550671610
- A. W. Anderson and N. G. Merckling, U. S. U.S. Patent 2,721,189 (页面存档备份,存于) (October 18, 1955)
- Stereospecific Homopolymerization of Cyclopentene Angewandte Chemie International Edition in English Volume 3, Issue 11, Date: November 1964, Pages: 723-729 G. Natta, G. Dall'Asta, G. Mazzanti doi:10.1002/anie.196407231
- Olefin Disproportionation. A New Catalytic Process R. L. Banks and G. C. Bailey Ind. Eng. Chem. Prod. Res. Dev.; 1964; 3(3) pp 170 - 173; doi:10.1021/i360011a002
- Olefin metathesis - A novel reaction for skeletal transformations of unsaturated hydrocarbons Tetrahedron Letters, Volume 8, Issue 34, 1967, Pages 3327-3329 Nissim Calderon, Hung Yu Chen and Kenneth W. Scott doi:10.1016/S0040-4039(01)89881-6
- Olefin metathesis. I. Acyclic vinylenic hydrocarbons Nissim Calderon, Eilert A. Ofstead, John P. Ward, W. Allen Judy, and Kenneth W. Scott J. Am. Chem. Soc. 1968; 90(15); 4133-4140. doi:10.1021/ja01017a039
- Catalyse de transformation des oléfines par les complexes du tungstène. II. Télomérisation des oléfines cycliques en présence d'oléfines acycliques Die Makromolekulare Chemie Volume 141, Issue 1, Date: 9 February 1971, Pages: 161-176 Par Jean-Louis Hérisson, Yves Chauvin doi:10.1002/macp.1971.021410112
- E. O. Fischer, A. Maasböl. . Angewandte Chemie International Edition in English. 1964, 3 (8): 580–581. doi:10.1002/anie.196405801.
- A proposed mechanism for the metal-catalysed disproportionation reaction of olefins Tetrahedron Letters, Volume 12, Issue 11, 1971, Pages 789-793 Glenn S. Lewandos and R. Pettit doi:10.1016/S0040-4039(01)96558-X
- Mechanism of the metal-catalyzed disproportionation of olefins Glenn S. Lewandos, R. Pettit J. Am. Chem. Soc. 1971; 93(25); 7087-7088. doi:10.1021/ja00754a067
- Possible intermediate in the tungsten-catalyzed olefin metathesis reaction Robert H. Grubbs, Terence K. Brunck J. Am. Chem. Soc.; 1972; 94(7); 2538-2540. doi:10.1021/ja00762a073
- Crystal structure of bis(triphenylphosphine)tetramethyleneplatinum(II) Carol G. Biefeld, Harry A. Eick, Robert H. Grubbs Inorg. Chem.; 1973; 12(9); 2166-2170. doi:10.1021/ic50127a046
- Mechanism of the olefin metathesis reaction Thomas J. Katz, James McGinnis J. Am. Chem. Soc.; 1975; 97(6); 1592-1594. doi:10.1021/ja00839a063
- Reactions of (diphenylcarbene)pentacarbonyltungsten(0) with alkenes. Role of metal-carbene complexes in cyclopropanation and olefin metathesis reactions Charles P. Casey, Terry J. Burkhardt J. Am. Chem. Soc.; 1974; 96(25); 7808-7809. doi:10.1021/ja00832a032
- Mechanism of the olefin metathesis reaction Robert H. Grubbs, Patrick L. Burk, Dale D. Carr J. Am. Chem. Soc. 1975; 97(11); 3265-3267. doi:10.1021/ja00844a082
- Consideration of the mechanism of the metal catalyzed olefin metathesis reaction Robert H. Grubbs, D. D. Carr, C. Hoppin, P. L. Burk J. Am. Chem. Soc. 1976; 98(12); 3478-3483. doi:10.1021/ja00428a015
- Olefin homologation with titanium methylene compounds F. N. Tebbe, G. W. Parshall, G. S. Reddy J. Am. Chem. Soc. 1978; 100(11); 3611-3613. doi:10.1021/ja00479a061
- Titanium metallacarbene-metallacyclobutane reactions: stepwise metathesis T. R. Howard, J. B. Lee, R. H. Grubbs J. Am. Chem. Soc. 1980; 102(22); 6876-6878. doi:10.1021/ja00542a050
- Synthesis of (+-)Δ9,12-capnellene using titanium reagents John R. Stille, Robert H. Grubbs J. Am. Chem. Soc. 1986; 108(4); 855-856. doi:10.1021/ja00264a058
- Titanacyclobutanes derived from strained, cyclic olefins: the living polymerization of norbornene Laura R. Gilliom, Robert H. Grubbs J. Am. Chem. Soc. 1986; 108(4); 733-742. doi:10.1021/ja00264a027
- Ring-opening polymerization of norbornene by a living tungsten alkylidene complex R. R. Schrock, J. Feldman, L. F. Cannizzo, R. H. Grubbs Macromolecules; 1987; 20(5); 1169-1172. doi:10.1021/ma00171a053
- Coordinated polymerization of the bicyclo-[2.2.1]-heptene-2 ring system (norbornene) in polar media Journal of Polymer Science Part A: General Papers Volume 3, Issue 3, Date: March 1965, Pages: 895-905 Francis W. Michelotti, William P. Keaveney doi:10.1002/pol.1965.100030305
- The ring opening metathesis polymerization of 7-oxabicyclo[2.2.1]hept-5-ene derivatives: a new acyclic polymeric ionophore Bruce M. Novak, Robert H. Grubbs J. Am. Chem. Soc. 1988; 110(3); 960-961. doi:10.1021/ja00211a043
- Ring-opening metathesis polymerization (ROMP) of norbornene by a Group VIII carbene complex in protic media SonBinh T. Nguyen, Lynda K. Johnson, Robert H. Grubbs, Joseph W. Ziller J. Am. Chem. Soc. 1992; 114(10); 3974-3975. doi:10.1021/ja00036a053
- Syntheses and activities of new single-component, ruthenium-based olefin metathesis catalysts SonBinh T. Nguyen, Robert H. Grubbs, Joseph W. Ziller J. Am. Chem. Soc. 1993; 115(21); 9858-9859. doi:10.1021/ja00074a086
- A Series of Well-Defined Metathesis Catalysts-Synthesis of [RuCl2(CHR)(PR3)2] and Its Reactions Angewandte Chemie International Edition in English Volume 34, Issue 18, Date: October 2, 1995, Pages: 2039-2041 Peter Schwab, Marcia B. France, Joseph W. Ziller, Robert H. Grubbs doi:10.1002/anie.199520391
- Synthesis and Applications of RuCl2(=CHR')(PR3)2: The Influence of the Alkylidene Moiety on Metathesis Activity Peter Schwab, Robert H. Grubbs, and Joseph W. Ziller J. Am. Chem. Soc. pp 100 - 110; 1996 (Article) doi:10.1021/ja952676d
- Pentamethyl complexes of niobium and tantalum R. R. Schrock, P. Meakin J. Am. Chem. Soc.; 1974; 96(16); 5288-5290. doi:10.1021/ja00823a064
- Preparation and characterization of tantalum(III) olefin complexes and tantalum(V) metallacyclopentane complexes made from acyclic .alpha. olefins S. J. McLain, C. D. Wood, R. R. Schrock J. Am. Chem. Soc. 1979; 101(16); 4558-4570. doi:10.1021/ja00510a022
- Preparation and characterization of active niobium, tantalum and tungsten metathesis catalysts Journal of Molecular Catalysis, Volume 8, Issues 1-3, May 1980, Pages 73-83 Richard Schrock, Scott Rocklage, Jeffrey Wengrovius, Gregory Rupprecht and Jere Fellmann doi:10.1016/0304-5102(80)87006-4
- Multiple metal-carbon bonds. 16. Tungsten-oxo alkylidene complexes as olefins metathesis catalysts and the crystal structure of W(O)(CHCMe3(PEt3)Cl2 Jeffrey H. Wengrovius, Richard R. Schrock, Melvyn Rowen Churchill, Joseph R. Missert, Wiley J. Youngs J. Am. Chem. Soc. 1980; 102(13); 4515-4516. doi:10.1021/ja00533a035
- Synthesis of molybdenum imido alkylidene complexes and some reactions involving acyclic olefins Richard R. Schrock, John S. Murdzek, Gui C. Bazan, Jennifer Robbins, Marcello DiMare, Marie O'Regan J. Am. Chem. Soc. 1990; 112(10); 3875-3886. doi:10.1021/ja00166a023
- Living ring-opening metathesis polymerization of 2,3-difunctionalized 7-oxanorbornenes and 7-oxanorbornadienes by Mo(CHCMe2R)(NC6H3-iso-Pr2-2,6)(O-tert-Bu)2 and Mo(CHCMe2R)(NC6H3-iso-Pr2-2,6)(OCMe2CF3)2 Guillermo C. Bazan, John H. Oskam, Hyun Nam Cho, Lee Y. Park, Richard R. Schrock J. Am. Chem. Soc. 1991; 113(18); 6899-6907. doi:10.1021/ja00018a028
- Synthesis of chiral molybdenum ROMP initiators and all-cis highly tactic poly(2,3-(R)2norbornadiene) (R = CF3 or CO2Me) David H. McConville, Jennifer R. Wolf, Richard R. Schrock J. Am. Chem. Soc. 1993; 115(10); 4413-4414. doi:10.1021/ja00063a090
延伸阅读
- . Chemical & Engineering News. 2002, 80 (51): 29–33 [2008-05-23]. (原始内容存档于2008-10-05).
- . Chemical & Engineering News. 2002, 80 (51): 34–38 [2008-05-23]. (原始内容存档于2008-10-05).
- Schrock, R. R. . Acc. Chem. Res. 1990, 23 (5): 158–165. doi:10.1021/ar00173a007.
- Schrock, R. R.; Hoveyda, A. H. . Angew. Chem. Int. Ed. 2003, 42 (38): 4592–4633. doi:10.1002/anie.200300576.
- Trnka, T. M.; Grubbs, R. H. . Acc. Chem. Res. 2001, 34 (1): 18–29. doi:10.1021/ar000114f.
- Grubbs, R. H.; Chang, S. . Tetrahedron. 1998, 54 (18): 4413–4450. doi:10.1016/S0040-4020(97)10427-6.
- Grubbs, R. H. . Tetrahedron. 2004, 60 (34): 7117–7140. doi:10.1016/j.tet.2004.05.124.
外部链接
- 金属卡宾络合物催化的烯烃复分解反应
- 烯烃复分解反应—有机化学.org (页面存档备份,存于)
- 有机合成中的复分解方法的发展 (页面存档备份,存于)
- 烯烃复分解反应(Olefin metathesis) (页面存档备份,存于)