抗肿瘤药
抗肿瘤药(英語:)也称为抗癌药、抗恶性肿瘤药,是指治疗恶性肿瘤的药物。[1]此类药物通过多种途径杀灭或抑制癌细胞来达到治疗恶性肿瘤的目的。根据药理作用的不同可以将抗肿瘤药分为细胞毒性药物和非细胞毒性药物,前者以DNA毒性药物为主,后者以分子靶向抗肿瘤药物为主。[2]常用的抗肿瘤药有:顺铂、多柔比星、紫杉醇、伊马替尼等。
| 抗肿瘤药 | |
|---|---|
| 药物种类 | |
 | |
| 用途 | 治疗恶性肿瘤 |
| 生物靶标 | 杀灭或抑制癌细胞 |
| ATC代码 | L01 |
| 外部链接 | |
| MeSH | E02.183.750.500 |
| AHFS/Drugs.com | 药物分类 |
传统的细胞毒性药物由于对癌细胞缺乏足够的选择性,在杀伤癌细胞的同时,对正常的组织细胞也产生不同程度的损伤作用。而随着肿瘤分子生物学和转化医学的发展,抗肿瘤药已从传统的细胞毒性药物向非细胞毒性药物发展。非细胞毒性药物具有高选择性和高治疗指数的特点,临床优势明显。[3]
用途
抗肿瘤药主要用于医疗方面,用来治疗癌症。[4]由于一些抗肿瘤药兼有抗病毒活性,所以它们也被用于治疗一些病毒性传染病。[5]某些甾体激素药物(内分泌治疗药),虽然无抗肿瘤活性但可以调节体内激素平衡,对某些功能性腺癌有抑制作用,因此常用于抗肿瘤药物的联合治疗。[3]同时抗肿瘤药也被运用于科学研究。人们通过对一些抗肿瘤药药理作用的研究,来进一步了解癌症的分子生物学特征。[3]
历史
第一个抗肿瘤药氮芥是由L.S.古德曼和A.Z.吉尔曼于20世纪40年代对芥子气(学名二氯二乙硫醚)的化学结构进行修饰而得到的,随后盐酸氮芥(Chlorethamine Hydrochloride)作为最早使用于临床的抗肿瘤药,于1949年被批准上市,用于治疗淋巴瘤和何杰金氏病。[1]而第一个含芳香基的氮芥类药物苯丁酸氮芥于1957年被批准上市,用于治疗慢性淋巴细胞白血病。[6]
早期的抗肿瘤新药大多是随机筛选,通过动物移植性肿瘤寻找的。肿瘤细胞中磷酰胺酶活性高于正常细胞,同时磷酰基作为吸电子基团能降低氮芥中氮原子上电子云的密度,基于这一思路,H.阿诺德于1957年合成了环磷酰胺,并在临床上取得成功。[7]同年,查尔斯·海德尔伯格等人根据电子等排原理合成了5-氟尿嘧啶,同样在临床上取得成功。[8]这两种药物也是首先根据理论而合成的有效抗肿瘤药物。[4]
20世纪初,保罗·埃尔利希提出了“魔弹”的构想,即期望某些特异性化合物能作为靶向分子将药物带至病灶部位,进而减少对正常组织或细胞的损伤,这是靶向制剂的最初构想。1948年,D.普莱斯曼与G.凯特利提出用抗体作为细胞生长抑制剂和放射性核素的载体,这是抗肿瘤药靶向制剂及单克隆抗体类抗肿瘤药的雏形。[9]1951年,W.H.贝尔沃尔特用碘-131标记的抗体治疗甲状腺肿瘤。[10]1958年,乔治·马特等将抗体连接到甲氨蝶呤上用以治疗白血病。1972年,T.高斯等将苯丁酸氮芥连接到抗体上治疗黑色素瘤。[11]以上这些试验充分验证了以抗体为抗肿瘤药或载体的可行性,但这些试验所使用的抗体均为多克隆抗体,专一性不理想,故效果有限。1975年,乔治斯·克勒与色萨·米尔斯坦发明了单克隆抗体技术。由于单克隆抗体的高度专一性,抗肿瘤药的靶向制剂开始以单克隆抗体为载体不断发展,同时也出现了许多单克隆抗体类抗肿瘤药。[12]
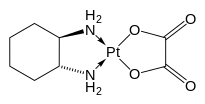
金属铂配合物的抗肿瘤生物活性研究起于20世纪60年代,当时美国生理学家巴内特·卢森堡等人在研究电磁场作用下微生物的生长情况时,发现在氯化铵介质中的铂电极周围大肠杆菌停止分裂繁殖。经研究确认顺-二氯·二氨合铂(Ⅱ)和顺-四氯·二氨合铂(Ⅳ)对细胞繁殖有抑制作用。随后,卢森堡及其合作者用患有肉瘤-180和白血病L1210的小鼠做实验,发现顺铂具有抗癌活性,最终使顺铂于1971年进入临床实验。[13][14][15]1978年FDA批准顺铂为睾丸癌和卵巢癌的治疗药。第二代金属铂配合物药物卡铂于20世纪80年代上市,而第一个手性金属铂配合物药物奥沙利铂于1996年上市。[1]

1962年,M.E.沃尔和M.C.瓦尼开始进行红豆杉树皮抗肿瘤活性成分的研究。沃尔负责短叶红豆杉(Taxus Breviolia)树皮中抗肿瘤活性成分的提取,他于1967年从树皮中分离得到了紫杉醇,收率仅0.014%。而瓦尼则使用沃尔提取的紫杉醇制备单晶,通过单晶X射线衍射技术于1971年确定了紫杉醇的化学结构。[16]1979年,生物学家S.B.霍维茨发现紫杉醇的靶点是微管蛋白。[17]1984年,美国国家癌症研究所进行了紫杉醇的一期临床实验,实验结果表明紫杉醇对于乳腺癌和卵巢癌具有非常好的疗效。[5]1989年,佛罗里达州立大学的罗伯特·霍尔顿教授从浆果紫杉的树叶中提取得到紫杉醇的前体10-去乙酰基巴卡亭Ⅲ(10-deacetylbaccatin Ⅲ,10-DBA),含量约0.1%,并以此进行半合成紫杉醇,解决了天然紫杉醇产量不足的问题。[18][19][20][16]
20世纪90年代末,汽巴-嘉基(1996年与山德士合并成诺华公司[21])通过靶向筛选的方式开发了最早的分子靶向抗肿瘤药物伊马替尼。[16]1998年6月,伊马替尼进入Ⅰ期临床实验,31例参与的患者在用药数周后白细胞计数即恢复正常,仅仅32个月后诺华公司就在全球范围递交了新药申请,美国FDA于2001年3月27日通过了给予其优先审批的资格。2001年5月10日伊马替尼在尚未完成Ⅲ期临床实验的情况下就被FDA批准提前上市,其审批进程比同类药物快了一倍。伊马替尼的研发成功开创了靶向性抗肿瘤药研发的新模式。[22]
分类
临床应用的抗肿瘤药种类较多且发展迅速,其分类迄今尚不完全统一,一般根据其药理作用和靶点进行以下分类。[1][23][24]
具体药物种类
| 中文名称(别称)[註 1] | 英文名称(别称) | 适应症及其他应用 | 作用机理 | 副作用 |
|---|---|---|---|---|
| 一.直接作用于DNA的药物(Drugs Directly Acting on DNA) | ||||
| 1.烷化剂(Alkylating Agents) | ||||
| 氮芥 | Chlorethamine | 淋巴瘤,何杰金氏病 | 氮芥类药物的氮原子碱性很强,在游离状态和生理pH时,易和β位氯原子作用生成高度活泼的氮丙啶离子,其为强亲电性的烷化剂,能与细胞成分的DNA、RNA或蛋白质中的亲核基团发生烷基化反应,形成交叉联结或脱嘌呤作用,使DNA链断裂,下一次复制时又可以使碱基配对错码,造成DNA结构或功能损坏。 | |
| 氧氮芥# | ||||
| 苯丁酸氮芥(瘤可宁) | Chlorambucil(Leukeran) | 慢性淋巴细胞白血病,淋巴瘤,何杰金氏病,卵巢癌等 | ||
| 美法仑(溶肉瘤素) | Melphalan | 卵巢癌,乳腺癌,淋巴瘤,多发性骨髓瘤等 | ||
| 乌拉莫司汀 | Uramustine | |||
| 氮甲(甲酰溶肉瘤素) | Formylmerphalan | 精原细胞瘤,淋巴瘤,多发性骨髓瘤等 | ||
| 环磷酰胺 | Cyclophosphamide(CTX) | 淋巴瘤,急性淋巴细胞白血病,多发性骨髓瘤,肺癌,神经母细胞瘤等 | ||
| 异环磷酰胺 | Ifosfamide(IFO) | 睾丸癌,淋巴瘤,肉瘤,膀胱癌等 | ||
| 氯磷酰胺 | Chlorophosphamide | 何杰金氏病,慢性淋巴细胞白血病等 | ||
| 噻替哌 | Thiotepa | 卵巢癌,乳腺癌,肝癌,膀胱癌等 | 与氮芥类药物作用相似,为氮芥类药物代谢后转变而成的活性中间体。 | |
| 丝裂霉素C | Mitomycin C | 各种腺癌如胃癌,乳腺癌,胰腺癌等 | ||
| 卡莫司汀(氯乙亚硝脲,卡氮芥) | Carmustine(BCNU) | 脑瘤,转移性肿瘤等 | 亚硝基脲类药物中,N-亚硝基的存在使得该氮原子与邻近羰基之间的键变得不稳定,在生理条件下分解生成亲电性基团,这些基团与DNA中的碱基和磷酸酯基发生烷基化反应。 | |
| 洛莫司汀(环己亚硝脲) | Lomustine(CCNU) | |||
| 司莫司汀(甲环亚硝脲) | Semustine(Me-CCNU) | 脑瘤,胃癌,肠癌,肺癌等 | ||
| 尼莫司汀 | Nimustine(ACNU) | 脑瘤,胃癌,肠癌,肺癌,何杰金氏病等 | ||
| 雷莫司汀 | Ranimustine | 胶质母细胞瘤,骨髓瘤,慢性骨髓性白血病,何杰金氏病等 | ||
| 链脲佐菌素(链左托星) | Streptozocin | 胰岛细胞瘤等 | ||
| 氯脲霉素 | Chlorozotocin | |||
| 白消安(马利兰) | Busulfan | 慢性粒细胞白血病,骨髓增殖性疾病等 | 与DNA中鸟嘌呤结合产生分子内交联,同时与氨基酸中的巯基发生双烷基化反应 | |
| 六甲蜜胺 | Altretamine | 卵巢癌,小细胞肺癌等的联合化疗 | 药物经代谢生成活性产物N-(甲基羟基)三聚氰胺,后者在细胞内进一步脱甲基生成亲电性基团,这些基团与DNA发生烷基化反应。 | |
| 丙卡巴肼(甲基苄肼) | Procarbazine | 何杰金氏病,骨髓瘤,黑色素瘤等 | 药物经代谢释放出甲基正离子并与DNA发生烷基化反应。 | |
| 达卡巴嗪 | Dacarbazine | 黑色素瘤,何杰金氏病等 | 药物经代谢释放出甲基正离子并与DNA发生烷基化反应,而其他的一些代谢产物则与嘌呤生物合成的中间产物结构相似,可干扰嘌呤的生物合成。 | |
| 曲贝替定 | Trabectedin(Yondelis) | 软组织肉瘤 | 为特殊烷化剂,作用于DNA双螺旋之间的沟槽,通过与DNA分子结合干扰细胞分裂与DNA修复,从而促进肿瘤细胞凋亡 | |
| 2.金属铂配合物(Platinum Complexes) | ||||
| 顺铂(顺氯铵铂) | Cisplatin(DDP) | 非精原细胞性睾丸癌,卵巢癌等,抗瘤谱广 | 铂类配合物进入肿瘤细胞后水解成水合物,该水合物在体内与DNA中的两个鸟嘌呤碱基N-7位络合形成一个封闭的五元螯合环,从而破坏了两条核苷酸链上嘌呤基和胞嘧啶之间的氢键,扰乱了DNA的正常双螺旋结构,使其局部变性失活而丧失复制能力。 | |
| 卡铂(碳铂) | Carboplatin(CBP) | |||
| 奥沙利铂 | Oxaliplatin | |||
| 奈达铂 | Nedaplatin | |||
| 3.博来霉素类(Bleomycin) | ||||
| 博来霉素 | Bleomycin(BLM) | 鳞状细胞癌(头颈,上消化道,生殖系统等),淋巴瘤联合治疗等 | 博来霉素类药物的化学结构的左边部分含有多个氨基酸、糖、嘧啶环及咪唑,右边部分含有平面的二噻唑环。在和DNA作用时,左边的部分和亚铁离子形成螯合物,从而激活药物并与DNA中胸腺嘧啶脱氧核苷酸的C-4'结合,使DNA缺损断裂;右边部分则与DNA的小沟中特定的部分结合导致DNA的裂解。 |
|
| 平阳霉素 | Pingyangmycin(PYM) | 鳞状细胞癌(头颈部),淋巴瘤联合治疗,乳腺癌等 | ||
| 4.DNA拓扑异构酶抑制剂(DNA Topoisomerase Inhibitors) | ||||
| 喜树碱 | Camptothecin(CPT) | 消化道肿瘤,肝癌,膀胱癌,白血病等 | 以喜树碱类药物为主,其化学结构中的含有β-羟基的内酯环和第一型拓扑异构酶反应,使DNA单链的断裂-再连接反应不能进行,从而抑制DNA转录,复制及细胞有丝分裂 | |
| 伊立替康 | Irinotecan(CPT-11) | 肺癌,结肠癌,卵巢癌,子宫癌,白血病等 | ||
| 拓扑替康 | Topotecan | 小细胞肺癌,肠癌,乳腺癌等 | ||
| 鲁比替康# | Rubitecan | 小细胞肺癌,肠癌,乳腺癌等 | ||
| 放线菌素D(更生霉素) | Dactinomycin(DACT) | 恶性葡萄胎,何杰金氏病,绒毛膜上皮癌,肾母细胞瘤 | 其平面结构中吩噁嗪酮母核与DNA结合,同时抑制第二型拓扑异构酶。 | |
| 多柔比星(阿霉素) | Doxorubicin(Adriamycin,ADM) | 耐药性急性淋巴细胞白血病,何杰金氏病,乳腺癌,胃癌等 | 药物结构中的蒽环或蒽醌嵌入到DNA的C-G碱基对之间,使DNA与第二型拓扑异构酶形成的复合物僵化,最终使DNA断裂。 | |
| 柔红霉素(柔毛霉素,红比霉素,正定霉素) | Daunorubicin(Daunomycin,rubidomycin,DRN) | |||
| 表柔比星(表阿霉素) | Epirubicin | |||
| 佐柔比星 | Zorubicin | |||
| 阿柔比星(阿克拉霉素) | Aclacinomicin A | |||
| 吡柔比星 | Pirarubicin | |||
| 氨苯吖啶(安吖啶) | Amsacrine(AMSA) | |||
| 米托蒽醌 | Mitoxantrone(NVT) | 晚期乳腺癌,非霍奇金氏淋巴瘤复发等 | ||
| 比生群(匹杉琼) | Pixantrone | |||
| 依托泊苷(鬼臼乙叉甙,足叶乙甙) | Etoposide(Vepesid,VP16) | 肺癌,睾丸癌 | 药物结构中的第4位通过差向异构化而得到的基团与第二型拓扑异构酶直接作用,从而阻止DNA复制及转录。 | |
| 替尼泊苷(鬼臼噻吩甙,特尼泊甙) | Teniposide(VM-26) | 肺癌,睾丸癌等 | ||
| 安萘菲特(苯二甲酰酯) | Amonafide(BIDA) | 小细胞肺癌 | 为第二型拓扑异构酶抑制剂,可选择性阻断DNA复制。 | |
| 二.干扰DNA合成的药物(Drugs Interfering with DNA Synthesis) | ||||
| 1.叶酸拮抗物(Folates Antimetabolites) | ||||
| 甲氨蝶呤 | Methotrexate(Amethopterine,MTX) | 急性白血病,绒毛膜上皮癌等 | 化学结构与二氢叶酸相似,作用于二氢叶酸还原酶,使二氢叶酸不能转化为四氢叶酸,从而影响辅酶F的生成,并干扰胸腺嘧啶核苷酸和嘌呤核苷酸的合成。 | |
| 氨基蝶呤(白血宁) | Aminopterin | |||
| 2.嘧啶拮抗物(Pyrimidine Antimetabolites) | ||||
| 5-氟尿嘧啶 | 5-Fluorouracil(5-FU) | 急性白血病,绒毛膜上皮癌等 | 此类药物在体内经代谢转变成5-氟尿嘧啶脱氧核苷酸,与胸腺嘧啶合成酶结合,再与辅酶5和10-次甲基四氢叶酸作用。由于C-F键稳定,导致靶细胞不能有效合成胸腺嘧啶脱氧核苷酸,从而抑制DNA合成。 | |
| 替加氟(呋氟尿嘧啶) | Tegafur(Ftorafur) | |||
| 双呋氟尿嘧啶# | Difuradin | |||
| 去氧氟尿苷 | Doxifluridine(5'-dFUR) | 胃癌,肠癌,乳腺癌等 | ||
| 卡莫氟 | Carmofur | |||
| 阿糖胞苷 | Cytarabine(Ara-C) | 急性粒细胞性白血病,单核细胞白血病等 | 与尿嘧啶衍生物类药物相似,抑制DNA聚合酶。 | |
| 伊诺他滨 | Enocitabine | |||
| 安西他滨(环胞苷) | Cyclocytidine | 各类急性白血病,抗单纯疱疹病毒(作为抗病毒药)等 | ||
| 吉西他滨 | Gemcitabine | 胰腺癌,中晚期小细胞肺癌等 | ||
| 地西他滨 | Decitabine | 各类急性白血病 | 抑制DNA甲基转移酶(DNMT) | |
| 3.嘌呤拮抗物(Purine Antimetabolites) | ||||
| 巯嘌呤 | Mercaptopurine(6-MP) | 急性淋巴细胞白血病的维持治疗,绒毛膜上皮癌等 | 此类药物经酶促转变为6-硫代次黄嘌呤核苷酸,抑制腺酰琥珀酸合成酶,阻止次黄嘌呤核苷酸转变为腺苷酸;同时抑制肌苷酸脱氢酶,阻止肌苷酸氧化为黄嘌呤核苷酸,从而抑制DNA和RNA的合成。 | |
| 磺巯嘌呤钠#(溶癌呤) | Sulfomercapine Sodium | |||
| 硫唑嘌呤#(依木兰) | Azathioprine(6-AP) | 白血病(现已停用),红斑狼疮,器官移植(作为免疫抑制剂)等 | ||
| 硫鸟嘌呤 | 6-Thioguanine(6-TG) | 白血病联合治疗等 | ||
| 喷司他丁 | Pentostatin | |||
| 氟达拉滨 | Fludarabine | 皮肤T细胞淋巴瘤,慢性淋巴细胞白血病,非霍奇金氏淋巴瘤等 | ||
| 克拉屈滨 | Cladribine | |||
| 奈拉滨 | Nelarabine | T细胞急性淋巴细胞白血病,T细胞淋巴瘤 | ||
| 4.多靶点拮抗物及其他抗代谢物 | ||||
| 雷替曲塞 | Raltitrexed | 晚期结肠癌等 | 同时具有叶酸拮抗物类药物及尿嘧啶衍生物类药物的作用。 | |
| 培美曲塞 | Pemetrexed | 非小细胞肺癌,耐药性间皮瘤 | ||
| 羟基脲 | Hydroxycarbamide(Hydroxyurea,HU) | 慢性粒细胞性白血病,头颈癌,卵巢癌等 | 抑制核苷酸还原酶,阻止胞苷酸转变为脱氧胞苷酸,从而抑制DNA合成,选择性作用于S期细胞。 | |
| 三.作用于结构蛋白的药物(Drugs Acting on Structurl Proteins) | ||||
| 1.抑制微管蛋白聚合的药物(Agents Inhibiting on Tubulin Polymerization) | ||||
| 秋水仙碱# | Colchicine | 乳腺癌(现已停用),痛风,類风湿性关节炎(作为免疫抑制剂)等 | 药物结构中的七元稠环与微管蛋白二聚体的α亚基和β亚基之间的位点结合,阻断细胞分裂。 | |
| 长春碱(长春花碱) | Vinblastin(VLB) | 各种实体瘤 | 药物结构中的二聚吲哚与未受损的微管蛋白在“生长末端”结合,同时药物在微管壁上有一低亲和力的位点,使微管在细胞内聚集形成聚集体,使肿瘤细胞停止于分裂中期。 | |
| 长春新碱 | Vincristine(VCR) | 儿童急性白血病等 | ||
| 长春地辛(长春酰胺) | Vindesine(VDS) | 急性淋巴细胞性白血病,慢性粒细胞性白血病等 | ||
| 长春瑞滨 | Vinorelbine(NRB) | 非小细胞肺癌等 | ||
| 2.抑制微管蛋白解聚的药物(Agents Inhibiting on Tubulin Depolymerization) | ||||
| 紫杉醇 | Taxtol(Paclitaxel) | 卵巢癌,乳腺癌,肺癌,黑色素瘤等 | 诱导并促使微管蛋白聚合,同时抑制所形成的微管束解聚,产生稳定的微管束,使微管束的动态再生受阻。 | |
| 多息紫杉醇(紫杉特尔,欧洲紫杉醇) | Docetaxel(Taxotere) | 除肾癌和肠癌外的其他实体瘤 | ||
| 3.干扰核蛋白体功能的药物 | ||||
| 三尖杉酯碱 | Harringtonine | 急性单核细胞白血病,慢性粒细胞白血病,何杰金氏病等 | 抑制蛋白质合成的起始阶段,使核蛋白体分解。 | |
| 高三尖杉酯碱 | Homoharringtonine | |||
| 4.影响氨基酸供应的药物 | ||||
| L-门冬酰胺酶 | L-Asparaginase | 急性淋巴细胞白血病联合治疗等 | 水解血清门冬酰胺,使癌细胞缺乏门冬酰胺供应,生长受到抑制。 |
|
| 四.小分子激酶抑制剂(Small-molecule Inhibitors of Kinases) | ||||
| 1.单靶点激酶抑制剂(Kinase Inhibitors of One Target) | ||||
| 伊马替尼(格列卫) | Imatinib(Glivec,Gleevec) | 费城染色体为阳性的慢性粒细胞白血病和胃肠道间质瘤 | 与Abl蛋白激酶ATP位点相结合,抑制激酶活性,阻止Bcr-Abl阳性细胞增殖并导致其凋亡。 | |
| 达沙替尼(扑瑞赛) | Dasatinib(Sprycel) | |||
| 尼罗替尼 | Nilotinib(Tasigna) | |||
| 波舒替尼 | Bosutinib | |||
| 泊那替尼 | Ponatinib | |||
| 吉非替尼(易瑞沙) | Gefitinib(Iressa) | 晚期或转移的非小细胞肺癌二线治疗 | 与细胞内激酶结构域结合,阻断EGFR(表皮生长因子受体酪氨酸激酶)的激酶活性及下游信号通路。 | |
| 厄洛替尼(特罗凯) | Erlotinib(Tarceva) | |||
| 埃克替尼 | Icotinib | |||
| 阿法替尼 | Afatinib | |||
| 坦罗莫司 | Temsirolimus(Torisel) | 晚期肾癌 | 阻断PI3K-Akt-mTOR信号通路和其他由mTOR介导的信号转导过程。 |
|
| 依维莫司 | Everolimus(Afinitor) | |||
| 维莫非尼 | Vemurafenib | BRAF V600E突变的不可切除或转移黑色素瘤 | 阻断B-Raf激酶 | |
| 达拉菲尼 | Dabrafenib | |||
| 依鲁替尼 | Ibrutinib | 套细胞淋巴瘤、慢性淋巴细胞白血病、巨球蛋白血症 | 阻断BTK蛋白酪氨酸激酶 | |
| 伊德利塞 | Idelalisib | 难治性急性淋巴细胞性白血病、难治性滤泡型B细胞非霍奇金氏淋巴瘤、难治性小淋巴细胞淋巴瘤 | 阻断PI3Kδ脂激酶 | |
| 奥沙替尼 | Osimertinib | 非小细胞肺癌 | 抑制EGFR蛋白酪氨酸激酶 | |
| 2.多靶点激酶抑制剂(Kinase Inhibitors of Multiple Targets) | ||||
| 索拉非尼(多吉美) | Sorafenib(Nexavar) | 肾癌、肝癌等 | 阻断Ras/Raf/MEK/ERK信号传导通路,同时抑制VEGFR(血管内皮生长因子受体)和PDGFR(血小板衍生生长因子受体)等受体酪氨酸激酶活性 | |
| 舒尼替尼(索坦) | Sunitinib(Sutent) | 晚期肾癌、胃肠道间质瘤、晚期胰腺癌 | 阻断VEGFR1/2/3和PDGFR细胞内酪氨酸激酶结构域的ATP结合部位,同时抑制c-kit(干细胞因子受体)、RET(胶质细胞源性神经营养因子受体)、CSF-1R(集落刺激因子受体-1)等其他蛋白酪氨酸激酶 | |
| 帕唑帕尼 | Pazopanib(Votrient) | 晚期肾癌、晚期软组织肉瘤 | 抑制VEGFR-1/2/3、PDGFR-α/β和c-kit蛋白酪氨酸激酶 | |
| 范得他尼 | Vandetanib(Zactima) | 晚期或转移的髓样甲状腺癌 | 抑制VEGFR、EGFR和RET蛋白酪氨酸激酶 | |
| 拉帕替尼 | Lapatinib(Tykerb) | 晚期或转移的乳腺癌 | 抑制ErbB1/EGFR和ErbB2/HER2蛋白酪氨酸激酶 | |
| 克唑替尼 | Crizotinib | ALK阳性转移性非小细胞肺癌 | 抑制ALK、C-MET和HGFR蛋白酪氨酸激酶 | |
| 鲁索利替尼 | Ruxolitinib | 中等或高度危险的骨髓纤维化 | 抑制JAK1和JAK2蛋白酪氨酸激酶 |
|
| 阿昔替尼 | Axitinib | 骨髓纤维化、真性红细胞增多症 | 抑制VEGFR、C-KIT、PDGFR等蛋白酪氨酸激酶 | |
| 瑞戈非尼 | Regorafenib | 转移性肠癌、晚期胃肠道间质细胞瘤 | 抑制VEGFR等蛋白酪氨酸激酶 | |
| 卡博替尼 | Cabozantinib | 进展或转移甲状腺髓样瘤 | 抑制VEGFR和C-MET蛋白酪氨酸激酶 | |
| 曲美替尼 | Trametinib | BRAF V600E突变的不可切除或转移黑色素瘤 | 抑制MEK1和MEK2丝氨酸、苏氨酸激酶 |
|
| 色瑞替尼 | Ceritinib | ALK阳性转移性非小细胞肺癌 | 抑制ALK等蛋白酪氨酸激酶 | |
| 帕布昔利布 | Palbociclib | 绝经期妇女ER阳性和HER2阴性的晚期乳腺癌 | 抑制CDK4和CDK6丝氨酸、苏氨酸激酶 |
|
| 乐伐替尼 | Lenvatinib | 局部复发或转移、进展和放疗难治性分化型甲状腺肿瘤、肝癌 | 抑制VEGFR、PDGFR等蛋白酪氨酸激酶 | |
| 五.其他抗肿瘤药物(Miscellaneous Anticancer Drugs) | ||||
| 1.蛋白酶体抑制剂(Proteasome Inhibitors) | ||||
| 硼替佐米(万珂) | Bortezomib(Velcade) | 多发性骨髓瘤 | 抑制蛋白酶体26S亚单位的糜蛋白酶和胰蛋白酶 | |
| 卡非佐米 | Carfilzomib | |||
| 2.组蛋白去乙酰化酶抑制剂(Histone Deacetylase Inhibitors) | ||||
| 伏立诺他(伏瑞斯特) | Vorinostat(SAHA) | 皮肤T细胞淋巴瘤 | 抑制组蛋白去乙酰化酶(HDAC)-1/2/3/6 |
|
| 3.单克隆抗体类药物(Monoclonal Antibody) | ||||
| 利妥昔单抗(美罗华) | Rituximab(Rituxan) | 非霍奇金氏淋巴瘤 | 与CD20抗原结合导致B淋巴细胞溶解 | |
| 阿仑珠单抗(坎帕斯) | Alemtuzumab(Campath) | 慢性淋巴性白血病 | 与CD52抗原结合导致带CD52抗原的靶细胞凋亡 | |
| 替依莫单抗(泽娃灵) | Ibritumomab(Zevalin) | 复发或难治的非霍奇金氏淋巴瘤 | 携带放射性同位素90Y,与CD20抗原结合,将同位素90Y富集于肿瘤部位,通过β射线杀死5mm范围内的肿瘤细胞 | |
| 托西莫单抗 | Tositumomab(Bexxar) | 非霍奇金氏淋巴瘤 | 携带放射性同位素131I,与CD20抗原结合,通过131I的放射性杀灭肿瘤细胞 | |
| 曲妥珠单抗(赫赛汀) | Trastuzumab(Herceptin) | HER-2(表皮生长因子受体)高表达的转移性乳腺癌 | 与HER-2(ErbB-2)选择性结合,阻断HER-2介导的PI3K和MAPK信号通路,抑制HER-2高表达的肿瘤细胞增殖 | |
| 西妥昔单抗(爱必妥) | Cetuximab(Erbitux) | 转移性肠癌、头颈部肿瘤 | 拮抗EGFR信号转导通路后抑制由该受体介导的肿瘤增殖 | |
| 帕尼单抗 | Panitumumab(Vectibix) | 转移性肠癌 | ||
| 尼妥珠单抗(泰欣生) | Nimotuzumab | HER-1阳性表达的Ⅲ/Ⅳ期鼻咽癌 | ||
| 帕妥珠单抗 | Pertuzumab(Perjeta) | HER-2阳性表达的乳腺癌 | ||
| 贝伐珠单抗(安维汀) | Bevacizumab(Avastin) | 转移性肠癌、晚期非小细胞肺癌、转移性肾癌、恶性胶质瘤 | 与VEGF(人血管内皮生长因子)结合,阻碍VEGF与其位于肿瘤血管内皮细胞上的受体(KDR和Flt-1)结合,抑制肿瘤血管生成 | |
| 依匹单抗 | Ipilimumab(Yervoy) | 黑色素瘤、肺癌 | 抑制CTLA4 |
|
| 派姆单抗(健痊得) | Pembrolizumab(Keytruda) | 黑色素瘤、非小细胞肺癌 | 抑制PD1 | |
| 4.调节体内激素平衡的药物[註 2] | ||||
| 己烯雌酚 | Diethylstilbestrol | 绝经期乳腺癌 | 调节体内激素平衡,抑制某些依赖激素生长的癌症,起辅助治疗的作用 | (参见泌尿生殖系统及性类固醇、体激素、糖皮质激素、类固醇皮质激素等) |
| 二甲基睾丸酮 | Methyltestosterone | 骨转移的晚期乳腺癌 | ||
| 丙酸睾丸酮 | Testosterone Propionate | |||
| 氟羟甲酮 | Fluoxymesterone | |||
| 甲羟孕酮酯(乙酸羟甲孕酮,甲孕酮,安宫黄体酮) | Medroxyprogesterone(MPA) | 乳腺癌、肾癌、子宫内膜癌 | ||
| 泼尼松 | Prednisone | 何杰金氏病和淋巴瘤的辅助治疗 | ||
| 他莫昔芬(三苯氧胺) | Tamoxifen(TAM) | 乳腺癌 | ||
| 戈舍瑞林 | Goserelin | 前列腺癌、绝经期的乳腺癌 | ||
| 亮丙瑞林 | Leuprorelin | 闭经前且雌激素受体阳性的前列腺癌和乳腺癌 | ||
| 氟他胺(氟硝丁酰胺) | Flutamide | 前列腺癌 | ||
| 托瑞米芬 | Toremifene | 绝经期的雌激素受体阳性转移性乳腺癌 | ||
| 来曲唑 | Letrozole | 绝经期的晚期乳腺癌 | ||
| 阿那曲唑 | Anastrozole | 绝经后乳腺癌的辅助治疗 | ||
| 氨鲁米特(氨基导眠能,氨格鲁米特,氨苯哌酮) | Aminoglutethimide(AG) | 绝经期的晚期乳腺癌 | ||
| 5.其他抗肿瘤机理的药物 | ||||
| 重组人血管内皮抑制素(恩度) | Endostar(Rh-Endostatin) | 非小细胞肺癌的辅助治疗 | 抑制肿瘤血管内皮细胞的增殖和迁移进而抑制肿瘤血管的生成 | |
| 维A酸(维甲酸) | Retinoic Acid(Tretinoin) | 急性早幼粒细胞白血病 | 调变并降解PML-RARa融合蛋白的维甲酸受体(RARα)的结构域,诱导白细胞分化成熟进而凋亡 |
|
| 亚砷酸(三氧化二砷) | Arsenious Acid(As2O3) | 急性早幼粒细胞白血病 | 调变并降解PML-RARa融合蛋白,下调bcl-2基因表达,诱导白细胞分化成熟进而凋亡 |
|
| 乌苯美司 | Ubenimex | 化疗或放疗的联合治疗,老年性免疫功能缺陷等 | 药物可竞争性抑制氨肽酶B及亮氨酸肽酶的活性,增强T淋巴细胞的功能,使NK细胞活性增强。同时可以促进集落刺激因子的合成从而刺激骨髓细胞的再生与分化。另外可以干扰肿瘤细胞代谢从而抑制肿瘤细胞增生。 | |
| 去甲斑螯素 | Norcantharidin | 肝癌、食管癌、胃癌等辅助化疗、肝硬化 | 抑制癌细胞蛋白质合成进而影响其DNA与RNA合成,同时降低癌激素(主要是环磷酸鸟苷-磷酸二酯酶)水平,并增加脾淋巴细胞产生白细胞介素Ⅱ、巨噬细胞产生白细胞介素Ⅰ的含量,从而提高机体免疫力 |
|
| 葫芦素B | Cucurbitacin B | 原发性肝癌的辅助治疗 | 具有保护肝等多种生物活性,同时能抑制STAT3转录因子活化,破坏肿瘤细胞的肌动蛋白细胞骨架 |
|
| 银杏叶提取物 | EGb761 | 转移性癌症的辅助治疗 | EGb761中含100多种化学成分,其中黄酮类和萜内酯类为有效成分,有抗肿瘤活性 |
|
作用机理
肿瘤细胞群包括增殖细胞群、静止细胞群(G0期)和无增殖能力细胞群。肿瘤增殖细胞群与全部肿瘤细胞群之比称生长比率(Growth Fraction,GF)。肿瘤细胞从一次分裂结束到下一次分裂结束的时间称为细胞周期,此间历经4个时相DNA合成前期(G0期)、DNA合成期(S期)、DNA合成后期(G2期)和有丝分裂期(M期)。[4]
细胞毒性药物
细胞毒性药物通过影响细胞周期的生化事件对不同周期的肿瘤细胞产生细胞毒性作用并延缓细胞周期的时相过渡。[25]依据药物对各周期或时相肿瘤细胞的敏感性不同,大致将细胞毒性药物分为两大类:
- 细胞周期非特异性药物(Cell Cycle Nonspecific Agents,CCNSA):能杀灭处于增殖周期各时相的细胞甚至包括G0期细胞的药物,如直接破坏DNA结构以及影响其复制或转录功能的药物(烷化剂、抗肿瘤抗生素及铂类配合物等)。此类药物对恶性肿瘤细胞的作用往往较强,能迅速杀死肿瘤细胞,其杀伤作用呈剂量依赖性,在机体能耐受的药物毒性限度内,作用随剂量的增加而成倍增强。[23]
- 细胞周期(时相)特异性药物(Cell Cycle Specific Agents,CCSA):仅对增殖周期的某些时相敏感而对G0期细胞不敏感的药物,如作用于S期细胞的抗代谢药物和作用于M期细胞的长春碱类药物。此类药物对肿瘤细胞的作用往往较弱,其杀伤作用呈时间依赖性,需要一定时间才能发挥作用,达到一定剂量后即使剂量再增加其作用不再增强。[23]
毒理学
目前临床使用的细胞毒性药物对肿瘤细胞和正常细胞尚缺乏理想的选择作用,即药物在杀伤恶性肿瘤细胞的同时,对某些正常的组织也有一定程度的损害,毒性反应成为化疗时使用剂量受到限制的关键因素,同时亦影响患者的生命质量。[27]非细胞毒性药物中的一些分子靶向药物(如肿瘤信号通路抑制剂)可以特异性的作用于肿瘤细胞的某些特定分子位点,而这些位点在正常细胞通常不表达或者很少表达。因此,非细胞毒性药物通常安全性高,耐受性好,毒性反应较轻。[28]
细胞毒性药物的不良反应

共有的毒性反应
- 骨髓抑制:是肿瘤化疗的最大障碍之一,除激素类、博来霉素和L-门冬酰胺酶外,大多数细胞毒性药物均有不同程度的骨髓抑制。骨髓造血细胞经化疗后外周血细胞数减少的几率决定于细胞的寿命,寿命越短的外周血细胞越容易减少,通常先出现白细胞减少,然后出现血小板降低,一般不会引起严重贫血。除了常用各种集落刺激因子如GM-CSF、G-CSF、M-CSF、EPO等来处理血细胞下降,护理中必须采取措施预防各种感染和防治出血等。[28]
- 消化道反应:是细胞毒性药物的最常见毒性反应。化疗引起的恶心、呕吐根据发生时间分为急性和迟发性两种类型。前者常发生在化疗后24小时内;后者发生在化疗24小时后。高度或中度致吐者可应用地塞米松和5-HT3受体拮抗剂(如昂丹司琼),轻度致吐者可应用甲氧氯普胺或氯丙嗪。另外化疗也可损害增殖活跃的消化道黏膜组织,容易引起口腔炎、口腔溃疡、舌炎、食管炎等,应注意口腔清洁卫生,防止感染。[28]
- 脱发:正常人头皮约有10万根头发,除其中10%~15%的生发细胞处于静止期外,其他大部分处于活跃生长,因此多数细胞毒性药物都能引起不同程度的脱发。在化疗时给患者戴上冰帽,使头皮冷却,局部血管痉挛,或止血带结扎于发际,减少药物到达毛囊而减轻脱发,停止化疗后头发仍可再生。[29]
特有的毒性反应
- 心脏毒性:以多柔比星最常见,可引起心肌退行性病变和心肌间质水肿。心脏毒性的发生可能与多柔比星诱导自由基生成有关。[28]
- 呼吸系统毒性:主要表现为间质性肺炎和肺纤维化,主要药物有博来霉素、卡莫斯汀、丝裂霉素C、甲氨蝶呤、吉非替尼等。长期大剂量使用博来霉素可引起间质性肺炎及肺纤维化,可能与肺内皮细胞缺少使博来霉素灭活的酶有关。[30]
- 肝脏毒性:部分细胞毒性药物如L-门冬酰胺酶、放线菌素D、环磷酰胺等可引起肝脏损害。[30]
- 泌尿系统毒性:大剂量环磷酰胺可引起出血性膀胱炎,可能与大量代谢物丙烯醛经泌尿道排泄有关,同时应用巯乙磺酸钠可预防其发生。顺铂由肾小管分泌,可损害近曲小管和远曲小管。保持充足的尿量有助减轻泌尿系统毒性。[29]
- 神经毒性:长春新碱最容易引起外周神经病变。顺铂、甲氨蝶呤和5-氟尿嘧啶偶尔也可引起一些神经毒性。[29]
- 过敏反应:凡属于多肽类化合物或蛋白质类的抗肿瘤药如L-门冬酰胺酶、博来霉素,静脉注射后容易引起过敏反应。紫杉醇的过敏反应可能与赋形剂聚氧乙基蓖麻油有关。[29]
- 组织坏死和深静脉血栓:刺激性强的药物如丝裂霉素C、多柔比星等可引起注射部位的血栓性静脉炎,注射液漏于血管外可致局部组织坏死,应避免注射不当。[27]
远期的毒性反应
非细胞毒性药物的不良反应
非细胞毒性药物毒性反应较轻,但仍然有一些副作用。[30]
单克隆抗体类药物
单克隆抗体类药物分为鼠源性单克隆抗体、嵌合单克隆抗体、人源化单克隆抗体和完全人源化单克隆抗体。其中,鼠源性单克隆抗体(各类以“莫单抗”(momab)为通用名尾缀的单克隆抗体类药物)特异性好,代谢快,但由于其不含人源化成分,会诱导人体产生人抗鼠抗体,因而副作用较大。[31]由于其较大的副作用,自2003年起再没有新的鼠源单克隆抗体药物进入临床研究。[22]嵌合单克隆抗体(各类以“昔单抗”(ximab)为通用名尾缀的单克隆抗体类药物)则是由鼠源性单克隆抗体的V区与人抗体的C区拼接而成,其人源成分占60%-70%,因而副作用有所降低,同时保留了其与抗原结合的特异性。[31]人源化单克隆抗体(各类以“组(珠)单抗/单抗”(zumab)为通用名尾缀的单克隆抗体类药物)则是将人抗体的CDR代之以鼠源性单克隆抗体的CDR,其人源成分约占90%,进一步减小了副作用,但与抗原的结合能力有所下降。[31]完全人源化单克隆抗体(各类以“木(人)单抗/单抗”(mumab/umab)为通用名尾缀的单克隆抗体类药物)是利用基因敲除技术将小鼠抗体基因敲除,代之以人抗体基因,后用抗原免疫小鼠,再经杂交瘤技术制备得来。由于其人源成分占100%,因而基本无副作用,同时治疗效果也不受影响。[31]
耐药性
肿瘤细胞对抗肿瘤药物产生耐药性是化疗失败的重要原因。[2]有些肿瘤细胞对某些抗肿瘤药具有天然耐药性(Natural Resistance),即肿瘤细胞对药物原来就不敏感的现象,如处于非增殖的G0期肿瘤细胞一般对多数抗肿瘤药不敏感。亦有的肿瘤细胞对于原来敏感的药物,治疗一段时间后才产生不敏感现象,称之为获得性耐药性(Acquired Resistance)。[32]其中表现最突出、最常见的耐药性是多药耐药性(Multiple Drug Resistance,MDR)或称多向耐药性(Pleiotropic Drug Resistance),即肿瘤细胞在接触一种抗肿瘤药后,产生了对多种结构不同、作用机制各异的其他抗肿瘤药的耐药性。[27]
耐药性产生的原因十分复杂,不同药物其耐药机制不同,同一种药物存在着多种耐药机制。耐药性的遗传学基础现已证明,肿瘤细胞在增殖过程中有较固定的突变率,每次突变均可导致耐药性瘤株的出现。因此,分裂次数愈多(亦即肿瘤愈大),耐药瘤株出现的机会愈大。肿瘤干细胞学说认为肿瘤干细胞的存在是导致肿瘤化疗失败的主要原因,耐药性是肿瘤干细胞的特性之一。[4]现代研究表明,肿瘤细胞更容易对分子靶向性药物产生耐药性。[27]
药剂学
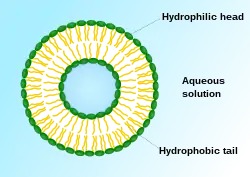
由于细胞毒性药物缺乏选择性,因而对机体有较大的副作用。[2]除了开发新型非细胞毒性药物以减少副作用,改变细胞毒性药物的剂型也是重要的手段之一。1906年,保罗·埃尔利希提出了靶向制剂(Targeting Drug System)的概念。靶向制剂为第四代药物剂型,被认为是抗肿瘤药的适宜剂型。[34]这类剂型不但增强了非细胞毒性药物的特异性,还使细胞毒性药物有了选择性。 早期靶向制剂以被动靶向制剂为主。[12]1961年,英国血液病专家A.D.班汉姆发明了脂质体。[35][36][37]1971年,脂质体首次被用作药物载体,这也是世界上最早的被动靶向制剂剂型。[38][39] 脂质体能使药物选择性地杀伤癌细胞或抑制癌细胞繁殖,增加药物对淋巴的选择性。由于肿瘤细胞中含有比正常细胞较高浓度的磷酸酶及酰酶,因此将抗癌药物包制成脂质体,不仅由于酶使药物容易释出,还使抗肿瘤药在靶区具有滞留性。[40][41]主动靶向制剂则包含了修饰的药物载体(如布洛芬锌微乳)、前体药物(如环磷酰胺)和药物大分子复合物。由于主动靶向制剂具有更高的选择性,能将药物定向地运送到靶区集中发挥药效,因而主动靶向制剂有更好的治疗效果。[12]
随着分子生物学的发展,对物理化学靶向制剂的研究也在不断深入。物理化学靶向制剂包含了磁性靶向制剂、栓塞靶向制剂、热敏靶向制剂、pH敏感靶向制剂等多种剂型。磁性靶向制剂是指将药物与铁磁性物质共同包裹于高分子聚合物载体中。当药物用于体内后,利用体外磁场的效应引导药物在体内定向移动和定位集中,主要用作抗癌药物载体。栓塞靶向制剂利用栓塞阻断对靶区的血供应和营养,使靶区的癌细胞缺血坏死。含有抗肿瘤药的栓塞制剂具有栓塞和靶向性化疗的双重作用。pH敏感制剂则利用肿瘤间质液的pH值比周围正常组织显著低的特点来达到靶向治疗的作用。[12]
制备途径
大多数抗肿瘤药通过全合成或半合成的方式进行工业制备,也有少数药物(如一些多肽类化合物或蛋白质类抗肿瘤药)是通过生物制药或天然成分提取的手段进行大规模生产。[1]
未来发展
随着在分子水平对肿瘤发病机制和细胞分化增殖和凋亡调控机制认识的深入,抗肿瘤药已从传统的细胞毒性作用向针对分子靶点的多环节作用的方向发展。[16]已经上市的新型分子靶向抗肿瘤药物可分为小分子化学药物和生物技术药物。前者主要由各种小分子激酶抑制剂组成,另外还包括蛋白酶体抑制剂和作用部分表观遗传学药物。后者以单克隆抗体类药物为代表,正逐渐成为抗肿瘤治疗的中坚力量。这些药物实际上超越了传统的直接细胞毒性药物。[6]当前分子靶向抗肿瘤药物的研发是新药开发领域的一个热点课题。[15]
基于靶点的新药研发
现今抗肿瘤药作用靶点的开发手段包括:从有效单体化合物着手发现靶点;以正常组织和病理组织基因表达差异发现靶点;通过定量分析和比较研究在正常和疾病状态下蛋白质的表达谱的改变发现靶点;以蛋白质相互作用为基础发现靶点;应用RNA干扰技术特异地抑制细胞中不同基因的表达,通过细胞的表型变化发现靶点等。[16]而新型抗肿瘤药的研发则是在靶点的三维结构基础上,利用计算机辅助药物设计的手段,从而快速筛选得到先导化合物,进而得到目标药物的过程。新型靶向性抗肿瘤药的研发靶点主要分为基因组与蛋白质组两种。[16]目前靶向性抗肿瘤药针对的驱动基因主要有两大类,一类是位于细胞膜上的受体分子(如HER2/neu等),另一类是位于细胞内重要信号通路的分子(如EGFR等)。由于基因发生插入、缺失、重排或扩增等突变导致驱动基因的活化,赋予了癌细胞的适应性,从而导致癌症的发生和发展。[22]而靶向性抗肿瘤药针对的蛋白质靶点则主要有疾病特异性蛋白(如多肽Op18、热激蛋白70等)、生物标记分子(如细胞角蛋白CK19等)、酶分子(如组蛋白去乙酰化酶(HDAC)等)等。[22]
脚注
- #:由于药效原因或毒性原因,已退出市场或变更用途的药物。
- 严格来说,此类药物属于内分泌治疗药物(ATC L02),但由于其在癌症治疗方面运用广泛,故将其列出。
参考文献
- 彭司勋; 尤启东. 第三版. 北京: 化学工业出版社. 2015-10: 487–521. ISBN 978-7-122-24850-3 (中文).
- 姜远英; 文爱东. 第四版. 北京: 人民卫生出版社. 2016-02. ISBN 978-7-117-22028-6 (中文).
- 李俊. 第五版. 北京: 人民卫生出版社. 2013-03. ISBN 978-7-117-16993-6 (中文).
- 王冠军; 赫捷. . 北京: 人民卫生出版社. 2013-03: 21–24,116–125. ISBN 978-7-117-17452-7 (中文).
- Patrick G. L. 5th. Oxford: Oxford Univercity Press. 2013: 514–578. ISBN 978-0-19-969739-7 (英语).
- 尤启东; 孙铁民. 第八版. 北京: 人民卫生出版社. 2016-02: 271-304. ISBN 978-7-117-22151-1 (中文).
- Arnold H.; Bourseaux F.; Brock N. . Nature. 1958, 181 (4613): 931. PMID 13526741.
- Heidelberger C.; Chaudhuri N. K.; Danneberg P.; et al. . Nature. March 1957, 179 (4561): 663–6. PMID 13418758. doi:10.1038/179663a0.
- Pressman D.; Keightley G. . J Immunol. 1948, 59 (2): 141–6. PMID 18864088.
- Freedberg A. S.; Ureles A. L.; Lesses M. F.; et al. . Am J Med. 1951, 11 (1): 44–54. PMID 14837925.
- Ghose T.; Norvell S. T.; Guclu A.; et al. . Eur J Cancer. 1975, 11 (5): 321–6. PMID 1171762.
- 方亮. 第八版. 北京: 人民卫生出版社. 2016-02. ISBN 978-7-117-22380-5 (中文).
- Rosenberg B.; Vancamp L.; Krigas T. . Nature. 1965, 205 (4972): 698–699. Bibcode:1965Natur.205..698R. PMID 14287410. doi:10.1038/205698a0.
- Rosenberg B.; Vancamp L.; Grimley E. B.; Thomson A. J. . J. Biol. Chem. March 1967, 242 (6): 1347–52. PMID 5337590.
- Lemke T. L.; Williams D. A. 7th. Baltimore: Lippincott Williams & Wilkins. 2011: 1199–1267. ISBN 978-1-609-133450 (英语).
- 方浩. 第三版. 北京: 人民卫生出版社. 2016-02. ISBN 978-7-117-21921-1 (中文).
- Horwitz S. B.; Goldman D. . Annual Review of Pharmacology and Toxicology. 2015, 55: 1–9 [2017-09-18]. doi:10.1146/annurev-pharmtox-010814-124519. (原始内容存档于2022-03-16).
- Robert A. Holton; Carmen Somoza; Hyeong Baik Kim; Feng Liang; et al. . J. Am. Chem. Soc. 1994, 116 (4). doi:10.1021/ja00083a066.
- Buchi G.; MacLeod William D.; Padilla J. . Journal of the American Chemical Society. 1964, 86 (20): 4438–4444. ISSN 0002-7863. doi:10.1021/ja01074a041.
- Büchi G.; Erickson R. E.; Wakabayashi Nobel. . Journal of the American Chemical Society. 1961, 83 (4): 927–938. ISSN 0002-7863. doi:10.1021/ja01465a042.
- Collins Glenn. . New York Times. 1996-03-07 [2017-09-22]. (原始内容存档于2017-08-08) (英语).
- 张景海. 第八版. 北京: 人民卫生出版社. 2016-02: 174–179. ISBN 978-7-117-22373-7 (中文).
- 杨宝峰; 苏定冯. 第八版. 北京: 人民卫生出版社. 2013-03: 438–456. ISBN 978-7-117-16975-2 (中文).
- 国家药典委员会. 2015年版. 北京: 中国医药科技出版社. 2015-08. ISBN 978-7-5067-4439-3 (中文).
- Hurley L. H. . Nature Reviews Cancer. 2002, 2 (3): 188–200. PMID 11990855. doi:10.1038/nrc749.
- Eck M.; Manley P. . Current Opinion in Cell Biology. 2009, 21 (2): 288–295. PMID 19217274. doi:10.1016/j.ceb.2009.01.014.
- Brunton L. L.; Lazo J. S. 11th. New York: The McGraw-Hill Companies Inc. 2011: 1021–1404. ISBN 978-0071422802 (英语).
- 楼宜嘉. 第四版. 北京: 人民卫生出版社. 2016-02. ISBN 978-7-117-22371-3 (中文).
- Katzung B. G. 12th. New York: The McGraw-Hill Companies Inc. 2012. ISBN 978-0071764018 (英语).
- 谭毓治; 唐圣松. 案例版. 北京: 科学出版社. 2009-12. ISBN 978-7-030-26292-9 (中文).
- 金伯泉; 曹雪涛. 第六版. 北京: 人民卫生出版社. 2013-03. ISBN 978-7-117-17101-4 (中文).
- Abraham D. J.; Rotella D. P. 7th. Hoboken: John Wiley & Sons Inc. 2010. ISBN 978-0470278154 (英语).
- Torchilin V. . Advanced Drug Delivery Reviews. 2006, 58 (14): 1532–55. PMID 17092599. doi:10.1016/j.addr.2006.09.009.
- Frank Heynick. . Hoboken: Ktav. 2002: 354–355. ISBN 0881257737 (英语).
- Bangham A. D.; Horne R. W. . Journal of Molecular Biology. 1964, 8 (5): 660–668. PMID 14187392. doi:10.1016/S0022-2836(64)80115-7.
- Horne R. W.; Bangham, A. D.; Whittaker V. P. . Nature. 1963, 200 (4913): 1340. PMID 14098499. doi:10.1038/2001340a0.
- Bangham A. D.; Horne R. W.; Glauert A. M.; et al. . Nature. 1962, 196: 952–955. PMID 13966357. doi:10.1038/196952a0.
- Cobleigh M.; Langmuir V. K.; Sledge G. W.; et al. . Seminars in Oncology. 2003, 30 (5 Suppl 16): 117–24. PMID 14613032. doi:10.1053/j.seminoncol.2003.08.013.
- Cobleigh M.; Langmuir V. K.; Sledge G. W.; et al. . Seminars in Oncology. 2003, 30 (5 Suppl 16): 117–24. PMID 14613032. doi:10.1053/j.seminoncol.2003.08.013.
- Lee J. H.; Yeo Yoon. . Chemical Engineering Science. Pharmaceutical Particles and Processing. 2015-03-24, 125: 75–84 [2017-09-18]. PMC 4322773
 . PMID 25684779. doi:10.1016/j.ces.2014.08.046. (原始内容存档于2022-06-22).
. PMID 25684779. doi:10.1016/j.ces.2014.08.046. (原始内容存档于2022-06-22). - Cho Kwangjae; Wang Xu; Nie Shuming; et al. . Clinical Cancer Research. 2008-03-01, 14 (5): 1310–1316. ISSN 1078-0432. PMID 18316549. doi:10.1158/1078-0432.CCR-07-1441.
- Hait W. N. . Nature Reviews Cancer. 2010, 9 (4): 253–4. PMID 20369394. doi:10.1038/nrd3144.