| Glycogen storage disease | |
|---|---|
| Other names | Glycogenosis, dextrinosis |
 | |
| Glycogen | |
| Specialty | Endocrinology |
A glycogen storage disease (GSD, also glycogenosis and dextrinosis) is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.[1]
GSD has two classes of cause: genetic and environmental. Genetic GSD is caused by any inborn error of carbohydrate metabolism (genetically defective enzymes or transport proteins) involved in these processes. In livestock, environmental GSD is caused by intoxication with the alkaloid castanospermine.[2]
However, not every inborn error of carbohydrate metabolism has been assigned a GSD number, even if it is known to affect the muscles or liver. For example, phosphoglycerate kinase deficiency (gene PGK1) has a myopathic form.
Also, Fanconi-Bickel syndrome (gene SLC2A2) and Danon disease (gene LAMP2) were declassed as GSDs due to being defects of transport proteins rather than enzymes; however, GSD-1 subtypes b, c, and d are due to defects of transport proteins (genes SLC37A4, SLC17A3) yet are still considered GSDs.
Phosphoglucomutase deficiency (gene PGM1) was declassed as a GSD due to it also affecting the formation of N-glycans; however, as it affects both glycogenolysis and glycosylation, it has been suggested that it should re-designated as GSD-XIV.[3]
(See inborn errors of carbohydrate metabolism for a full list of inherited diseases that affect glycogen synthesis, glycogen breakdown, or glucose breakdown.)
Types
| Type (Eponym) | Enzyme deficiency (Gene[4]) | Incidence (births) | Hypo- glycemia? | Hepato- megaly? | Hyper- lipidemia? | Muscle symptoms | Development/ prognosis | Other symptoms |
|---|---|---|---|---|---|---|---|---|
| GSD 0
(Lewis' disease)[5] | Glycogen synthase (Muscle GYS1 / Liver GYS2) | 1 in 20,000–25,000[6] | Liver 0a: Yes
Muscle 0b: No | No | No | (Muscle 0b) Glycogen deficiency in muscle fibres. Type I muscle fibre predominance. Exercise-induced, muscle fatigue, myalgia, fainting.[7][8] Occasional muscle cramping | (Liver 0a) Growth failure in some cases.[9]
(Muscle 0b) Risk of sudden death in childhood due to cardiac arrest.[7] |
(Liver 0a) Epilepsy[9] (Muscle 0b) Rarely epilepsy, tonic-clonic seizures.[7] Arrhythmia, long QT syndrome.[8] |
| GSD I / GSD 1 (von Gierke's disease) | Glucose-6-phosphatase / Glucose-6-phosphate translocase (G6PC / SLC37A4 /SLC17A3) | 1 in 50,000 – 100,000[10][11][12] | Yes | Yes | Yes | None | Growth failure | Lactic acidosis, hyperuricemia |
| GSD II / GSD 2 (Pompe disease, formerly GSD-IIa) Danon disease (formerly GSD-IIb) | Acid alpha-glucosidase
(GAA) Lysosome-associated membrane protein 2 (LAMP2) | Pompe disease is 1 in 13,000.[13] | No | Yes | No | Muscle weakness, exercise intolerance, abnormal lysosomal glycogen accumulation in muscle biopsy. Late-onset Pompe may have a pseudoathletic appearance of hypertrophic calf muscles.[14]
The symptoms of both Pompe and Danon diseases are very similar due to a defect in lysosomes. However, in Danon disease, some show abnormal glycogen accumulation, but not all.[15] | Progressive proximal skeletal muscle weakness with varied timeline to threshold of functional limitation (early childhood to adulthood). Approximately 15% of the Pompe population is classified as infantile Pompe which is typically deadly within the first year if untreated. | Heart failure (infantile), respiratory difficulty (due to muscle weakness) |
| GSD III / GSD 3 (Cori's disease or Forbes' disease) | Glycogen debranching enzyme (AGL) | 1 in 100,000 | Yes | Yes | Yes | Myopathy. May have a pseudoathletic appearance of hypertrophic muscles.[16] | Failure to thrive[17] |
myogenic hyperuricemia[18] |
| GSD IV / GSD 4 (Andersen's disease) | Glycogen branching enzyme (GBE1) | 1 in 500,000[19] | No | Yes, also cirrhosis | No | Myopathy and dilated cardiomyopathy | Failure to thrive, death at age ~5 years | |
| GSD V / GSD 5 (McArdle's disease) | Muscle glycogen phosphorylase (PYGM) | 1 in 100,000 – 500,000[20][19] | No | No | No | Exercise-induced muscle fatigue and cramps. Rhabdomyolysis possible. May have a pseudoathletic appearance of hypertrophic calf muscles.[21] | Renal failure by myoglobinuria, second wind phenomenon, inappropriate rapid heart rate (sinus tachycardia) response to exercise, myogenic hyperuricemia[18] | |
| GSD VI / GSD 6 (Hers' disease) | Liver glycogen phosphorylase (PYGL) | 1 in 65,000 – 85,000[22] | Yes | Yes | Yes[23] | None | initially benign, developmental delay follows. | |
| GSD VII / GSD 7 (Tarui's disease) | Muscle phosphofructokinase (PFKM) | 1 in 1,000,000[24] | No | No | No | Exercise-induced muscle cramps and weakness | developmental delay | In some haemolytic anaemia,
myogenic hyperuricemia[18] |
| GSD IX / GSD 9 | Phosphorylase kinase (PHKA2 / PHKB / PHKG2 / PHKA1) | ? | Yes | Yes | Yes | IXd Exercise-induced muscle cramps, stiffness, weakness (fatigue), and pain.[25] | Liver type: Delayed motor development, Developmental delay | |
| GSD X / GSD 10 | Muscle Phosphoglycerate mutase(PGAM2) | ? | ? | ? | ? | Exercise-induced muscle cramps and weakness[26] | Myoglobinuria[27] | |
| GSD XI / GSD 11 | Muscle lactate dehydrogenase (LDHA) | ? | ? | ? | ? |
Exercise-induced muscle cramps, stiffness, pain.[28] |
||
| Fanconi-Bickel syndrome formerly GSD XI / GSD 11, no longer considered a GSD | Glucose transporter (GLUT2) | ? | Yes |
Yes | No | None | ||
| GSD XII / GSD 12 (Aldolase A deficiency) | Aldolase A (ALDOA) | ? | No | In some | No | Exercise intolerance, cramps. In some Rhabdomyolysis. | Hemolytic anemia and other symptoms | |
| GSD XIII / GSD 13 | β-enolase (ENO3) | ? | No | ? | No | Exercise intolerance, cramps | Increasing intensity of myalgias over decades[29] | Serum CK: Episodic elevations; Reduced with rest[29] |
| CDG1T (formally GSD XIV / GSD 14) | Phosphoglucomutase-1(PGM1) | ? | Episodic | ? | No | Two forms: exclusively myopathic and multi-system (including muscles).[30]
Myopathy (including exercise-related fatigue, exercise intolerance, muscle weakness). Muscle biopsy shows glycogen accumulation.[31] |
Short stature, some have developmental delay, and rarely delayed puberty.[31] | Highly variable phenotype and severity. Commonly elevated serum CK, abnormal serum transferrin (loss of complete N-glycans), short stature, cleft palate, bifid uvula, and hepatopathy.[31]
Second Wind phenomenon in some[32] but not all[3] |
| GSD XV / GSD 15 | Glycogenin-1 (GYG1) | Rare[33] | No | No | No | Muscle atrophy, exercise intolerance, muscle biopsy shows abnormal glycogen depletion and marked proliferation of slow-twitch (type 1/oxidative) muscle fibres and mitochondrial proliferation. | Slowly progressive weakness over decades | Arrhythmia, biopsy of heart showed abnormal glycogen deposits (different from polyglucosan bodies) in cardiomyocytes.[34] |
Remarks:
- Some GSDs have different forms, e.g. infantile, juvenile, adult (late-onset).
- Some GSDs have different subtypes, e.g. GSD1a / GSD1b, GSD9A1 / GSD9A2 / GSD9B / GSD9C / GSD9D.[4]
- GSD type 0: Although glycogen synthase deficiency does not result in storage of extra glycogen in the liver, it is classified with the GSDs as type 0 because it is another defect of glycogen storage and can cause similar problems.
- GSD type VIII (GSD 8): In the past, liver phosphorylase-b kinase deficiency was considered a distinct condition,[35] however it has been classified with GSD type VI[22] and GSD IXa1;[36] it has been described as X-linked recessive inherited.[37] GSD IX has become the dominant classification for this disease, grouped with the other isoenzymes of phosphorylase-b kinase deficiency.[38]
- GSD type XI (GSD 11): Fanconi-Bickel syndrome (GLUT2 deficiency), hepatorenal glycogenosis with renal Fanconi syndrome, no longer considered a glycogen storage disease, but a defect of glucose transport.[4] The designation of GSD type XI (GSD 11) has been repurposed for muscle lactate dehydrogenase deficiency (LDHA).
- GSD type XIV (GSD 14): No longer classed as a GSD, but as a congenital disorder of glycosylation type 1T (CDG1T), affects the phosphoglucomutase enzyme (gene PGM1).[4] Phosphoglucomutase 1 deficiency is both a glycogenosis and a congenital disorder of glycosylation.[39] Individuals with the disease have both a glycolytic block as muscle glycogen cannot be broken down, as well as abnormal serum transferrin (loss of complete N-glycans).[39] As it affects glycogenolysis, it has been suggested that it should re-designated as GSD-XIV.[3]
- Lafora disease is considered a complex neurodegenerative disease and also a glycogen metabolism disorder.[40]
- Polyglucosan storage myopathies are associated with defective glycogen metabolism[41]
- (Not McArdle disease, same gene but different symptoms) Myophosphorylase-a activity impaired: Autosomal dominant mutation on PYGM gene. AMP-independent myophosphorylase activity impaired, whereas the AMP-dependent activity was preserved. No exercise intolerance. Adult-onset muscle weakness. Accumulation of the intermediate filament desmin in the myofibers of the patients.[42][43] Myophosphorylase comes in two forms: form 'a' is phosphorylated by phosphorylase kinase, form 'b' is not phosphorylated. Both forms have two conformational states: active (R or relaxed) and inactive (T or tense). When either form 'a' or 'b' are in the active state, then the enzyme converts glycogen into glucose-1-phosphate. Myophosphorylase-b is allosterically activated by AMP being in larger concentration than ATP and/or glucose-6-phosphate. (See Glycogen phosphorylase§Regulation).
- Unknown glycogenosis related to dystrophy gene deletion: patient has a previously undescribed myopathy associated with both Becker muscular dystrophy and a glycogen storage disorder of unknown aetiology.[44]
Diagnosis

Methods to diagnose glycogen storage diseases include history and physical examination for associated symptoms, blood tests for associated metabolic disturbances, and genetic testing for suspected mutations.[16][45] It may also include a non-ischemic forearm test, exercise stress test, or 12-minute walk test (12MWT).[45] Advancements in genetic testing are slowly diminishing the need for biopsy; however, in the event of a VUS and inconclusive exercise tests, a biopsy would then be necessary to confirm diagnosis.[45]
Differential diagnoses
Muscle
Glycogen storage diseases that involve skeletal muscle typically have exercise-induced (dynamic) symptoms, such as muscle fatigue, rather than fixed weakness (static) symptoms.[46] Differential diagnoses for glycogen storage diseases that involve fixed muscle weakness, particularly of the proximal muscles, would be an inflammatory myopathy or a limb-girdle muscular dystrophy.[46]
For those with exercise intolerance and/or proximal muscle weakness, the endocrinopathies should be considered.[47][48][49] The timing of the symptoms of exercise intolerance, such as muscle fatigue and cramping, is important in order to help distinguish it from other metabolic myopathies such as fatty acid metabolism disorders.[50]
Problems originating within the circulatory system, rather than the muscle itself, can produce exercise-induced muscle fatigue, pain and cramping that alleviates with rest, resulting from inadequate blood flow (ischemia) to the muscles. Ischemia that often produces symptoms in the leg muscles includes intermittent claudication, popliteal artery entrapment syndrome, and chronic venous insufficiency.
Diseases disrupting the neuromuscular junction can cause abnormal muscle fatigue, such as myasthenia gravis, an auto-immune disease.[51] Similar, are Lambert–Eaton myasthenic syndrome (auto-immune) and the congenital myasthenic syndromes (genetic).
Diseases can disrupt glycogen metabolism secondary to the primary disease. Abnormal thyroid function—hypo- and hyperthyroidism—can manifest as myopathy with symptoms of exercise-induced muscle fatigue, cramping, muscle pain and may include proximal weakness or muscle hypertrophy (particularly of the calves).[52][48] Hypothyroidism up-regulates glycogen synthesis and down-regulates glycogenolysis and glycolysis; conversely, hyperthyroidism does the reverse, up-regulating glycogenolysis and glycolysis while down-regulating glycogen synthesis.[53][54][55][48][56]
Prolonged hypo- and hyperthyroid myopathy leads to atrophy of type II (fast-twitch/glycolytic) muscle fibres, and a predominance of type I (slow-twitch/oxidative) muscle fibres.[54][48][49] Muscle biopsy shows abnormal muscle glycogen: high accumulation in hypothyroidism and low accumulation in hyperthyroidism.[56][53][54] Hypothyroid myopathy includes Kocher-Debre-Semelaigne syndrome (childhood-onset), Hoffman syndrome (adult-onset), myasthenic syndrome, and atrophic form.[56]
In patients with increased growth hormone, muscle biopsy includes, among other features, excess glycogen deposition.[57]
It is interesting to note, in comparison to hypothyroid myopathy, that McArdle disease (GSD-V), which is by far the most commonly diagnosed of the muscle GSDs and therefore the most studied,[58][45][59] has as its second highest comorbidity endocrine disease (chiefly hypothyroidism)[60][45] and that some patients with McArdle disease also have hypertrophy of the calf muscles.[21] Late-onset Pompe disease (GSD-II) also has calf hypertrophy and hyopthyroidism as comorbidities.[14][61][62]
Poor diet and malabsorption diseases (such as celiac disease) may lead to malnutrition of essential vitamins necessary for glycogen metabolism within the muscle cells. Malnutrition typically presents with systemic symptoms, but in rare instances can be limited to myopathy.[63] Vitamin D deficiency myopathy (also known as osteomalic myopathy due to the interplay between vitamin D and calcium) results in muscle weakness, predominantly of the proximal muscles; with muscle biopsy showing abnormal glycogen accumulation, atrophy of type II (fast-twitch/glycolytic) muscle fibres, and diminished calcium uptake by the sarcoplasmic reticulum (needed for muscle contraction).[64][65][66] Although Vitamin D deficiency myopathy typically includes muscle atrophy,[64] rarely calf muscle hypertrophy has been reported.[67][68]
Exercise-induced, electrically silent, muscle cramping and stiffness (transient muscle contractures or "pseudomyotonia") are seen not only in GSD types V, VII, IXd, X, XI, XII, and XIII, but also in Brody disease, Rippling muscle disease types 1 and 2, and CAV3-related hyperCKemia (Elevated serum creatine phosphokinase).[26] Unlike the other myopathies, in Brody disease the muscle cramping is painless.[69][70] Like GSD types II, III, and V, a pseudoathletic appearance of muscle hypertrophy is also seen in some with Brody disease and Rippling muscle disease.[69][71][72]
Erythrocyte lactate transporter defect (formerly Lactate transporter defect, myopathy due to) also includes exercise-induced, electrically silent, painful muscle cramping and transient contractures; as well as exercise-induced muscle fatigue.[26][73] EMG and muscle biopsy is normal however, as the defect is not in the muscle but in the red blood cells that should clear lactate buildup from exercising muscles.[73]
Although most muscular dystrophies have fixed muscle weakness rather than exercise-induced muscle fatigue and/or cramping, there are a few exceptions. Limb–girdle muscular dystrophy autosomal recessive 23 (LGMD R23) has calf hypertrophy and exercise-induced cramping.[74] Myofibrillar myopathy 10 (MFM10) has exercise-induced muscle fatigue, cramping and stiffness, with hypertrophic neck and shoulder girdle muscles.[75] LGMD R28 has calf hypertrophy and exercise-induced muscle fatigue and pain.[76] LGMD R8 has calf pseudohypertrophy and exercise-induced weakness (fatigue) and pain.[77] LGMD R15 (a.k.a MDDGC3) has muscle hypertrophy, proximal muscle weakness, and muscle fatigue.[78]
DMD-related myopathies of Duchenne and Becker muscular dystrophy are known for fixed muscle weakness and pseudohypertrophic calf muscles, but they also have secondary muscular mitochondrial impairment causing low ATP production; as well as decreasing type II (fast-twitch/glycolytic) muscle fibres, producing a predominance of type I (slow-twitch/oxidative) muscle fibres.[79] DMD-related childhood-onset milder phenotypes present with exercise-induced muscle cramping, stiffness, pain, fatigue, and elevated CK.[80] Becker muscular dystrophy has adult-onset exercise-induced muscle cramping, pain, and elevated CK.[81]
Tubular aggregate myopathy (TAM) types 1 and 2 has exercise-induced muscle pain, fatigue, stiffness, with proximal muscle weakness and calf muscle pseudohypertrophy. TAM1 has cramping at rest, while TAM2 has cramping during exercise.[82][83][84][85] Stormorken syndrome includes the symptoms of TAM, but is a more severe presentation including short stature and other abnormalities.[83] Satoyoshi syndrome has exercise-induced painful muscle cramps, muscle hypertrophy, and short stature.[86] Dimethylglycine dehydrogenase deficiency has muscle fatigue, elevated CK, and fishy body odour.[87] Myopathy with myalgia, increased serum creatine kinase, with or without episodic rhabdomyolysis (MMCKR) has exercise-induced muscle cramps, pain, and fatigue; with some exhibiting proximal muscle weakness.[88]
Liver
(help wikipedia by contributing to this subsection)
Treatment
Treatment is dependent on the type of glycogen storage disease. Von Gierke disease (GSD-I) is typically treated with frequent small meals of carbohydrates and cornstarch, called modified cornstarch therapy, to prevent low blood sugar, while other treatments may include allopurinol and human granulocyte colony stimulating factor.[89]
Cori/Forbes disease (GSD-III) treatment may use modified cornstarch therapy, a high protein diet with a preference to complex carbohydrates. However, unlike GSD-I, gluconeogenesis is functional, so simple sugars (sucrose, fructose, and lactose) are not prohibited.[16]
A ketogenic diet has demonstrated beneficial for McArdle disease (GSD-V) as ketones readily convert to acetyl CoA for oxidative phosphorylation, whereas free fatty acids take a few minutes to convert into acetyl CoA.[90][91]
For phosphoglucomutase deficiency (formerly GSD-XIV), D-galactose supplements and exercise training has shown favourable improvement of signs and symptoms.[30] In terms of exercise training, some patients with phosphoglucomutase deficiency also experience "second wind."[30][32]
For McArdle disease (GSD-V), regular aerobic exercise utilizing "second wind" to enable the muscles to become aerobically conditioned, as well as anaerobic exercise (strength training) that follows the activity adaptations so as not to cause muscle injury, helps to improve exercise intolerance symptoms and maintain overall health.[45][59][92][93] Studies have shown that regular low-moderate aerobic exercise increases peak power output, increases peak oxygen uptake (VO2peak), lowers heart rate, and lowers serum CK in individuals with McArdle disease.[92][93]
Regardless of whether the patient experiences symptoms of muscle pain, muscle fatigue, or cramping, the phenomenon of second wind having been achieved is demonstrable by the sign of an increased heart rate dropping while maintaining the same speed on the treadmill.[93] Inactive patients experienced second wind, demonstrated through relief of typical symptoms and the sign of an increased heart rate dropping, while performing low-moderate aerobic exercise (walking or brisk walking).[93]
Conversely, patients that were regularly active did not experience the typical symptoms during low-moderate aerobic exercise (walking or brisk walking), but still demonstrated second wind by the sign of an increased heart rate dropping.[93][94] For the regularly active patients, it took more strenuous exercise (very brisk walking/jogging or bicycling) for them to experience both the typical symptoms and relief thereof, along with the sign of an increased heart rate dropping, demonstrating second wind.[93][94][95]
In young children (<10 years old) with McArdle disease (GSD-V), it may be more difficult to detect the second wind phenomenon. They may show a normal heart rate, with normal or above normal peak cardio-respiratory capacity (VO2max).[45][96] That said, patients with McArdle disease typically experience symptoms of exercise intolerance before the age of 10 years,[45] with the median symptomatic age of 3 years.[58][97]
Tarui disease (GSD-VII) patients do not experience the "second wind" phenomenon; instead are said to be "out-of-wind."[45][59][98] However, they can achieve sub-maximal benefit from lipid metabolism of free fatty acids during aerobic activity following a warm-up.[45]
Epidemiology
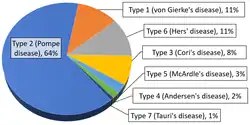
Overall, according to a study in British Columbia, approximately 2.3 children per 100,000 births (1 in 43,000) have some form of glycogen storage disease.[99] In the United States, they are estimated to occur in 1 per 20,000–25,000 births.[10] Dutch incidence rate is estimated to be 1 per 40,000 births. While a Mexican incidence showed 6.78:1000 male newborns.[12][100]
Within the category of muscle glycogenoses (muscle GSDs), McArdle disease (GSD-V) is by far the most commonly diagnosed.[58]
See also
References
- ↑ Cantú-Reyna, C.; Santos-Guzmán, J.; Cruz-Camino, H.; Vazquez Cantu, D.L.; Góngora-Cortéz, J.J.; Gutiérrez-Castillo, A. (2019). "Glucose-6-Phosphate dehydrogenase deficiency incidence in a Hispanic population". Journal of Neonatal-Perinatal Medicine. 12 (2): 203–207. doi:10.3233/NPM-1831. PMID 30741698. S2CID 73452760.
- ↑ Stegelmeier BL, Molyneux RJ, Elbein AD, James LF (May 1995). "The lesions of locoweed (Astragalus mollissimus), swainsonine, and castanospermine in rats". Veterinary Pathology. 32 (3): 289–98. doi:10.1177/030098589503200311. PMID 7604496. S2CID 45016726.
- 1 2 3 Stojkovic, Tanya; Vissing, John; Petit, François; Piraud, Monique; Orngreen, Mette C.; Andersen, Grete; Claeys, Kristl G.; Wary, Claire; Hogrel, Jean-Yves; Laforêt, Pascal (2009-07-23). "Muscle Glycogenosis Due to Phosphoglucomutase 1 Deficiency". New England Journal of Medicine. 361 (4): 425–427. doi:10.1056/NEJMc0901158. ISSN 0028-4793. PMID 19625727.
- 1 2 3 4 "Glycogen Metabolism". Themedicalbiochemistrypage.org. 29 April 2020. Retrieved 5 July 2022.
- ↑ "Glycogen Storage Diseases". Cleveland Clinic. Retrieved 2023-12-29.
- ↑ "Glycogen-Storage Disease Type 0 (GSD-0) (Glycogen Synthetase Deficiency): Background, Pathophysiology, Epidemiology". 2022-10-10.
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 3 "GLYCOGEN STORAGE DISEASE 0, MUSCLE; GSD0B". www.omim.org. Retrieved 2023-12-29.
- 1 2 "Glycogen Storage Disease type 0" (PDF). MedLine Plus.
- 1 2 "GLYCOGEN STORAGE DISEASE 0, LIVER; GSD0A". www.omim.org. Retrieved 2023-12-29.
- 1 2 eMedicine Specialties > Glycogen-Storage Disease Type I Author: Karl S Roth. Updated: Aug 31, 2009
- ↑ "Glycogen Storage Disease Type I". Association for Glycogen Storage Diseases (AGSD). October 2006. Archived from the original on 11 April 2021.
- 1 2 Cantú-Reyna, C.; Santos-Guzmán, J.; Cruz-Camino, H.; .Vazquez Cantu, D.L.; Góngora-Cortéz, J.J.; Gutiérrez-Castillo, A. (4 February 2019). "Glucose-6-Phosphate dehydrogenase deficiency incidence in a Hispanic population". Journal of Neonatal-Perinatal Medicine. 12 (2): 203–207. doi:10.3233/NPM-1831. PMID 30741698. S2CID 73452760.
- ↑ Bodamer, Olaf A.; Scott, C. Ronald; Giugliani, Roberto; Pompe Disease Newborn Screening Working Group (2017). "Newborn Screening for Pompe Disease". Pediatrics. 140 (Suppl 1): S4–S13. doi:10.1542/peds.2016-0280C. PMID 29162673. S2CID 43782810.
- 1 2 Menon, M. Suraj; Roopch, P. Sreedharan; Kabeer, K. Abdulkhayar; Shaji, C. Velayudhan (July 2016). "Calf Muscle Hypertrophy in Late Onset Pompe's Disease". Archives of Medicine and Health Sciences. 4 (2): 251. doi:10.4103/2321-4848.196188. ISSN 2321-4848. S2CID 58424073.
- ↑ "OMIM - # 300257 DANON DISEASE". OMIM — Online Medelian Inheritance in Man. 3 June 1986.
- 1 2 3 Kishnani, Priya S.; Austin, Stephanie L.; Arn, Pamela; Bali, Deeksha S.; Boney, Anne; Case, Laura E.; Chung, Wendy K.; Desai, Dev M.; El-Gharbawy, Areeg; Haller, Ronald; Smit, G. Peter A.; Smith, Alastair D.; Hobson-Webb, Lisa D.; Wechsler, Stephanie Burns; Weinstein, David A. (July 2010). "Glycogen Storage Disease Type III diagnosis and management guidelines". Genetics in Medicine. 12 (7): 446–463. doi:10.1097/GIM.0b013e3181e655b6. ISSN 1530-0366. PMID 20631546. S2CID 4609175.
- ↑ Tegay, David H (March 15, 2022). "Genetics of Glycogen-Storage Disease Type III Clinical Presentation: History, Physical, Causes". Medscape Reference. Retrieved October 24, 2023.
- 1 2 3 Mineo I, Kono N, Hara N, Shimizu T, Yamada Y, Kawachi M, Kiyokawa H, Wang YL, Tarui S. Myogenic hyperuricemia. A common pathophysiologic feature of glycogenosis types III, V, and VII. N Engl J Med. 1987 Jul 9;317(2):75-80. doi: 10.1056/NEJM198707093170203. PMID 3473284.
- 1 2 Stuart, Grant; Ahmad, Nargis (2011). "Perioperative care of children with inherited metabolic disorders". Continuing Education in Anaesthesia, Critical Care & Pain. 11 (2): 62–68. doi:10.1093/bjaceaccp/mkq055.
- ↑ Khattak, Zoia E.; Ashraf, Muddasir (January 2022). McArdle Disease. Treasure Island, Florida (FL): StatPearls Publishing. PMID 32809620. Archived from the original on 27 April 2022. Retrieved 7 July 2022.
{{cite book}}:|work=ignored (help) - 1 2 Rodríguez-Gómez, I.; Santalla, A.; Díez-Bermejo, J.; Munguía-Izquierdo, D.; Alegre, L. M.; Nogales-Gadea, G.; Arenas, J.; Martín, M. A.; Lucía, A.; Ara, I. (November 2018). "Non-osteogenic muscle hypertrophy in children with McArdle disease". Journal of Inherited Metabolic Disease. 41 (6): 1037–1042. doi:10.1007/s10545-018-0170-7. hdl:10578/19657. ISSN 1573-2665. PMID 29594644. S2CID 4394513.
- 1 2 Blenda, Anna V.; Chosed, Renee J.; Windle, Mary L.; Descartes, Maria; Curto, Lynne I; Kaye, Edward (4 Aug 2008). "Genetics of Glycogen Storage Disease Type VI (Hers Disease)". eMedicine (Medscape Reference). Archived from the original on 1 January 2022.
- ↑ Goldman, Lee; Schafer, Andrew (2012). Goldman's Cecil medicine (24th ed.). Philadelphia: Elsevier/Saunders. p. 1356. ISBN 978-1-4377-1604-7.
- ↑ "Rare Disease Database". Orpha.net. Retrieved 2015-09-20.
- ↑ "GLYCOGEN STORAGE DISEASE IXd; GSD9D". www.omim.org. Retrieved 2023-12-29.
- 1 2 3 "Exercise-induced muscle cramps (Concept Id: C1855578) - MedGen - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2023-12-28.
- ↑ Reference, Genetics Home. "Phosphoglycerate mutase deficiency". Genetics Home Reference. Retrieved 2019-02-06.
- ↑ "GLYCOGEN STORAGE DISEASE XI; GSD11". www.omim.org. Retrieved 2023-12-29.
- 1 2 "Glycogenoses".
- 1 2 3 Altassan, Ruqaiah; Radenkovic, Silvia; Edmondson, Andrew C.; Barone, Rita; Brasil, Sandra; Cechova, Anna; Coman, David; Donoghue, Sarah; Falkenstein, Kristina; Ferreira, Vanessa; Ferreira, Carlos; Fiumara, Agata; Francisco, Rita; Freeze, Hudson; Grunewald, Stephanie (January 2021). "International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management". Journal of Inherited Metabolic Disease. 44 (1): 148–163. doi:10.1002/jimd.12286. ISSN 0141-8955. PMC 7855268. PMID 32681750.
- 1 2 3 "Entry - #614921 - CONGENITAL DISORDER OF GLYCOSYLATION, TYPE It; CDG1T — OMIM". omim.org. Retrieved 2023-02-23.
- 1 2 Preisler, Nicolai; Cohen, Jonathan; Vissing, Christoffer Rasmus; Madsen, Karen Lindhardt; Heinicke, Katja; Sharp, Lydia Jane; Phillips, Lauren; Romain, Nadine; Park, Sun Young; Newby, Marta; Wyrick, Phil; Mancias, Pedro; Galbo, Henrik; Vissing, John; Haller, Ronald Gerald (November 2017). "Impaired glycogen breakdown and synthesis in phosphoglucomutase 1 deficiency". Molecular Genetics and Metabolism. 122 (3): 117–121. doi:10.1016/j.ymgme.2017.08.007. PMID 28882528.
- ↑ Malfatti E, Nilsson J, Hedberg-Oldfors C, Hernandez-Lain A, Michel F, Dominguez-Gonzalez C, Viennet G, Akman HO, Kornblum C, Van den Bergh P, Romero NB, Engel AG, DiMauro S, Oldfors A (2014) A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Ann Neurol 76(6):891-898
- ↑ Moslemi, Ali-Reza; Lindberg, Christopher; Nilsson, Johanna; Tajsharghi, Homa; Andersson, Bert; Oldfors, Anders (April 2010). "Glycogenin-1 Deficiency and Inactivated Priming of Glycogen Synthesis". New England Journal of Medicine. 362 (13): 1203–1210. doi:10.1056/NEJMoa0900661. ISSN 0028-4793. PMID 20357282.
- ↑ Ludwig M, Wolfson S, Rennert O (October 1972). "Glycogen storage disease, type 8". Arch. Dis. Child. 47 (255): 830–833. doi:10.1136/adc.47.255.830. PMC 1648209. PMID 4508182.
- ↑ GLYCOGEN STORAGE DISEASE IXa1; GSD9A1 OMIM — Online Mendelian Inheritance in Man
- ↑ "Definition: glycogen storage disease type VIII from Online Medical Dictionary". Archived from the original on 2009-07-03. Retrieved 2008-09-01.
- ↑ Herbert, Mrudu; Goldstein, Jennifer L.; Rehder, Catherine; Austin, Stephanie; Kishnani, Priya S.; Bali, Deeksha S. (1993), Adam, Margaret P.; Everman, David B.; Mirzaa, Ghayda M.; Pagon, Roberta A. (eds.), "Phosphorylase Kinase Deficiency", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 21634085, retrieved 2023-02-26
- 1 2 Tegtmeyer, Laura C.; Rust, Stephan; van Scherpenzeel, Monique; Ng, Bobby G.; Losfeld, Marie-Estelle; Timal, Sharita; Raymond, Kimiyo; He, Ping; Ichikawa, Mie; Veltman, Joris; Huijben, Karin; Shin, Yoon S.; Sharma, Vandana; Adamowicz, Maciej; Lammens, Martin (2014-02-06). "Multiple Phenotypes in Phosphoglucomutase 1 Deficiency". New England Journal of Medicine. 370 (6): 533–542. doi:10.1056/NEJMoa1206605. ISSN 0028-4793. PMC 4373661. PMID 24499211.
- ↑ Ortolano S, Vieitez I et al. Loss of cortical neurons underlies the neuropathology of Lafora disease. Mol Brain 2014;7:7 PMC 3917365
- ↑ Hedberg-Oldfors C, Oldfors A. Polyglucosan storage myopathies. Mol Aspects Med. 2015 Dec;46:85-100. doi: 10.1016/j.mam.2015.08.006. Epub 2015 Aug 13. PMID 26278982.
- ↑ Echaniz-Laguna A, Lornage X, Laforêt P, Orngreen MC, Edelweiss E, Brochier G, Bui MT, Silva-Rojas R, Birck C, Lannes B, Romero NB, Vissing J, Laporte J, Böhm J. A New Glycogen Storage Disease Caused by a Dominant PYGM Mutation. Ann Neurol. 2020 Aug;88(2):274-282. doi: 10.1002/ana.25771. Epub 2020 Jun 3. PMID 32386344.
- ↑ Echaniz-Laguna, A.; Lornage, X.; Edelweiss, E.; Laforêt, P.; Eymard, B.; Vissing, J.; Laporte, J.; Böhm, J. (October 2019). "O.5A new glycogen storage disorder caused by a dominant mutation in the glycogen myophosphorylase gene (PYGM)". Neuromuscular Disorders. 29: S39. doi:10.1016/j.nmd.2019.06.023. S2CID 203582211.
- ↑ Rose MR, Howard RS, Genet SA, McMahon CJ, Whitfield A, Morgan-Hughes JA. A case of myopathy associated with a dystrophin gene deletion and abnormal glycogen storage. Muscle Nerve. 1993 Jan;16(1):57-62. doi: 10.1002/mus.880160110. PMID 8423832.
- 1 2 3 4 5 6 7 8 9 10 Lucia, Alejandro; Martinuzzi, Andrea; Nogales-Gadea, Gisela; Quinlivan, Ros; Reason, Stacey; International Association for Muscle Glycogen Storage Disease study group (December 2021). "Clinical practice guidelines for glycogen storage disease V & VII (McArdle disease and Tarui disease) from an international study group". Neuromuscular Disorders. 31 (12): 1296–1310. doi:10.1016/j.nmd.2021.10.006. ISSN 1873-2364. PMID 34848128. S2CID 240123241.
- 1 2 Darras, B. T.; Friedman, N. R. (February 2000). "Metabolic myopathies: a clinical approach; part I". Pediatric Neurology. 22 (2): 87–97. doi:10.1016/s0887-8994(99)00133-2. ISSN 0887-8994. PMID 10738913.
- ↑ Bhavaraju-Sanka, R.; Jackson, C. E. (2014-01-01), "Myopathy, Endocrine", in Aminoff, Michael J.; Daroff, Robert B. (eds.), Encyclopedia of the Neurological Sciences (Second Edition), Oxford: Academic Press, pp. 259–264, ISBN 978-0-12-385158-1, retrieved 2023-05-24
- 1 2 3 4 Rodolico, Carmelo; Bonanno, Carmen; Pugliese, Alessia; Nicocia, Giulia; Benvenga, Salvatore; Toscano, Antonio (2020-09-01). "Endocrine myopathies: clinical and histopathological features of the major forms". Acta Myologica. 39 (3): 130–135. doi:10.36185/2532-1900-017. ISSN 1128-2460. PMC 7711326. PMID 33305169.
- 1 2 Sharma, Vikas; Borah, Papori; Basumatary, Lakshya J.; Das, Marami; Goswami, Munindra; Kayal, Ashok K. (2014). "Myopathies of endocrine disorders: A prospective clinical and biochemical study". Annals of Indian Academy of Neurology. 17 (3): 298–302. doi:10.4103/0972-2327.138505. ISSN 0972-2327. PMC 4162016. PMID 25221399.
- ↑ Bhai, Salman (September 2021). "Neuromuscular Notes: Diagnosing Metabolic Myopathies". Practical Neurology. Retrieved May 24, 2023.
- ↑ Gilhus, Nils Erik (2021-03-01). "Physical training and exercise in myasthenia gravis". Neuromuscular Disorders. 31 (3): 169–173. doi:10.1016/j.nmd.2020.12.004. hdl:11250/2767222. ISSN 0960-8966. S2CID 229372884.
- ↑ "Myopathies associated with thyroid disease". MedLink Neurology. Retrieved 2023-05-24.
- 1 2 Dimitriadis, G D; Leighton, B; Parry-Billings, M; West, D; Newsholme, E A (1989-01-15). "Effects of hypothyroidism on the sensitivity of glycolysis and glycogen synthesis to insulin in the soleus muscle of the rat". Biochemical Journal. 257 (2): 369–373. doi:10.1042/bj2570369. ISSN 0264-6021. PMC 1135589. PMID 2649073.
- 1 2 3 Celsing, F.; Blomstrand, E.; Melichna, J.; Terrados, N.; Clausen, N.; Lins, P. E.; Jansson, E. (April 1986). "Effect of hyperthyroidism on fibre-type composition, fibre area, glycogen content and enzyme activity in human skeletal muscle". Clinical Physiology. 6 (2): 171–181. doi:10.1111/j.1475-097x.1986.tb00066.x. ISSN 0144-5979. PMID 2937605.
- ↑ Brenta, Gabriela (2011-09-19). "Why Can Insulin Resistance Be a Natural Consequence of Thyroid Dysfunction?". Journal of Thyroid Research. 2011: e152850. doi:10.4061/2011/152850. ISSN 2090-8067. PMC 3175696. PMID 21941681.
- 1 2 3 Fariduddin, Maria M.; Bansal, Nidhi (2023), "Hypothyroid Myopathy", StatPearls, Treasure Island (FL): StatPearls Publishing, PMID 30137798, retrieved 2023-05-24
- ↑ Orrell, Richard W. (2007-01-01), "Endocrine myopathies", Myopathies, Handbook of Clinical Neurology, vol. 86, Elsevier, pp. 343–355, doi:10.1016/S0072-9752(07)86017-9, ISBN 9780444518996, PMID 18809009, retrieved 2023-05-24
- 1 2 3 Reason, S. L.; Voermans, N.; Lucia, A.; Vissing, J.; Quinlivan, R.; Bhai, S.; Wakelin, A. (2023-06-12). "Development of Continuum of Care for McArdle disease: A practical tool for clinicians and patients". Neuromuscular Disorders. 33 (7): 575–579. doi:10.1016/j.nmd.2023.05.006. ISSN 1873-2364. PMID 37354872. S2CID 259141690.
- 1 2 3 Wakelin, Andrew (2017). Living With McArdle Disease (PDF). IamGSD (Internation Association for Muscle Glycogen Storage Disease).
- ↑ Scalco, Renata S.; Lucia, Alejandro; Santalla, Alfredo; Martinuzzi, Andrea; Vavla, Marinela; Reni, Gianluigi; Toscano, Antonio; Musumeci, Olimpia; Voermans, Nicol C.; Kouwenberg, Carlyn V.; Laforêt, Pascal; San-Millán, Beatriz; Vieitez, Irene; Siciliano, Gabriele; Kühnle, Enrico (2020-11-24). "Data from the European registry for patients with McArdle disease and other muscle glycogenoses (EUROMAC)". Orphanet Journal of Rare Diseases. 15 (1): 330. doi:10.1186/s13023-020-01562-x. ISSN 1750-1172. PMC 7687836. PMID 33234167.
- ↑ Schneider, Joseph; Burmeister, Lynn A.; Rudser, Kyle; Whitley, Chester B.; Jarnes Utz, Jeanine (September 2016). "Hypothyroidism in late-onset Pompe disease". Molecular Genetics and Metabolism Reports. 8: 24–27. doi:10.1016/j.ymgmr.2016.06.002. ISSN 2214-4269. PMC 4932620. PMID 27408821.
- ↑ Fatehi, Farzad; Ashrafi, Mahmoud Reza; Babaee, Marzieh; Ansari, Behnaz; Beiraghi Toosi, Mehran; Boostani, Reza; Eshraghi, Peyman; Fakharian, Atefeh; Hadipour, Zahra; Haghi Ashtiani, Bahram; Moravej, Hossein; Nilipour, Yalda; Sarraf, Payam; Sayadpour Zanjani, Keyhan; Nafissi, Shahriar (2021). "Recommendations for Infantile-Onset and Late-Onset Pompe Disease: An Iranian Consensus". Frontiers in Neurology. 12: 739931. doi:10.3389/fneur.2021.739931. ISSN 1664-2295. PMC 8490649. PMID 34621239.
- ↑ Rasheed, Khalid; Sethi, Pooja; Bixby, Eric (May 2013). "Severe vitamin d deficiency induced myopathy associated with rhabydomyolysis". North American Journal of Medical Sciences. 5 (5): 334–336. doi:10.4103/1947-2714.112491. ISSN 2250-1541. PMC 3690793. PMID 23814767.
- 1 2 Polly, Patsie; Tan, Timothy C. (2014). "The role of vitamin D in skeletal and cardiac muscle function". Frontiers in Physiology. 5: 145. doi:10.3389/fphys.2014.00145. ISSN 1664-042X. PMC 3995052. PMID 24782788.
- ↑ Yoshikawa, S.; Nakamura, T.; Tanabe, H.; Imamura, T. (June 1979). "Osteomalacic myopathy". Endocrinologia Japonica. 26 (Suppl): 65–72. doi:10.1507/endocrj1954.26.supplement_65. ISSN 0013-7219. PMID 467350.
- ↑ Das, Anamica; Gopinath, Suchitra D.; Arimbasseri, Gopalakrishnan Aneeshkumar (February 2022). "Systemic ablation of vitamin D receptor leads to skeletal muscle glycogen storage disorder in mice". Journal of Cachexia, Sarcopenia and Muscle. 13 (1): 467–480. doi:10.1002/jcsm.12841. ISSN 2190-6009. PMC 8818613. PMID 34877816.
- ↑ Hassan, Ijas; Bhanudeep, Singanamalla; Madaan, Priyanka; Chhajed, Monika; Saini, Lokesh (2021). "Bilateral Calf Hypertrophy and Isolated Motor Delay: Think Beyond Muscular Dystrophy". Journal of Pediatric Neurosciences. 16 (2): 173–174. doi:10.4103/jpn.JPN_171_20. ISSN 1817-1745. PMC 8706592. PMID 35018192.
- ↑ Reimers, C. D.; Schlotter, B.; Eicke, B. M.; Witt, T. N. (November 1996). "Calf enlargement in neuromuscular diseases: a quantitative ultrasound study in 350 patients and review of the literature". Journal of the Neurological Sciences. 143 (1–2): 46–56. doi:10.1016/s0022-510x(96)00037-8. ISSN 0022-510X. PMID 8981297. S2CID 25971689.
- 1 2 "BRODY DISEASE; BROD". www.omim.org. Retrieved 2023-12-28.
- ↑ Molenaar, Joery P.; Verhoeven, Jamie I.; Rodenburg, Richard J.; Kamsteeg, Erik J.; Erasmus, Corrie E.; Vicart, Savine; Behin, Anthony; Bassez, Guillaume; Magot, Armelle; Péréon, Yann; Brandom, Barbara W.; Guglielmi, Valeria; Vattemi, Gaetano; Chevessier, Frédéric; Mathieu, Jean (2020-02-01). "Clinical, morphological and genetic characterization of Brody disease: an international study of 40 patients". Brain: A Journal of Neurology. 143 (2): 452–466. doi:10.1093/brain/awz410. ISSN 1460-2156. PMC 7009512. PMID 32040565.
- ↑ "RIPPLING MUSCLE DISEASE 1; RMD1". www.omim.org. Retrieved 2023-12-28.
- ↑ "RIPPLING MUSCLE DISEASE 2; RMD2". www.omim.org. Retrieved 2023-12-28.
- 1 2 "ERYTHROCYTE LACTATE TRANSPORTER DEFECT". www.omim.org. Retrieved 2023-12-28.
- ↑ "MUSCULAR DYSTROPHY, LIMB-GIRDLE, AUTOSOMAL RECESSIVE 23; LGMDR23". www.omim.org. Retrieved 2023-12-28.
- ↑ "MYOFIBRILLAR MYOPATHY 10; MFM10". www.omim.org. Retrieved 2023-12-28.
- ↑ "MUSCULAR DYSTROPHY, LIMB-GIRDLE, AUTOSOMAL RECESSIVE 28; LGMDR28". www.omim.org. Retrieved 2023-12-28.
- ↑ "MUSCULAR DYSTROPHY, LIMB-GIRDLE, AUTOSOMAL RECESSIVE 8; LGMDR8". www.omim.org. Retrieved 2023-12-28.
- ↑ "MUSCULAR DYSTROPHY-DYSTROGLYCANOPATHY (LIMB-GIRDLE), TYPE C, 3; MDDGC3". www.omim.org. Retrieved 2023-12-28.
- ↑ Heydemann, Ahlke (2018-06-20). "Skeletal Muscle Metabolism in Duchenne and Becker Muscular Dystrophy-Implications for Therapies". Nutrients. 10 (6): 796. doi:10.3390/nu10060796. ISSN 2072-6643. PMC 6024668. PMID 29925809.
- ↑ Veerapandiyan, Aravindhan; Shashi, Vandana; Jiang, Yong-Hui; Gallentine, William Brian; Schoch, Kelly; Smith, Edward Clinton (December 2010). "Pseudometabolic presentation of dystrophinopathy due to a missense mutation". Muscle & Nerve. 42 (6): 975–979. doi:10.1002/mus.21823. ISSN 1097-4598. PMC 5506871. PMID 21104870.
- ↑ "MUSCULAR DYSTROPHY, BECKER TYPE; BMD". www.omim.org. Retrieved 2023-12-29.
- ↑ "Tubular aggregate myopathy - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Retrieved 2023-11-11.
- 1 2 Morin, Gilles; Biancalana, Valérie; Echaniz-Laguna, Andoni; Noury, Jean-Baptiste; Lornage, Xavière; Moggio, Maurizio; Ripolone, Michela; Violano, Raffaella; Marcorelles, Pascale; Maréchal, Denis; Renaud, Florence; Maurage, Claude-Alain; Tard, Céline; Cuisset, Jean-Marie; Laporte, Jocelyn (January 2020). "Tubular aggregate myopathy and Stormorken syndrome: Mutation spectrum and genotype/phenotype correlation". Human Mutation. 41 (1): 17–37. doi:10.1002/humu.23899. ISSN 1098-1004. PMID 31448844.
- ↑ "MYOPATHY, TUBULAR AGGREGATE, 1; TAM1". www.omim.org. Retrieved 2023-11-11.
- ↑ "MYOPATHY, TUBULAR AGGREGATE, 2; TAM2". www.omim.org. Retrieved 2023-11-11.
- ↑ "SATOYOSHI SYNDROME". www.omim.org. Retrieved 2023-12-28.
- ↑ "DIMETHYLGLYCINE DEHYDROGENASE DEFICIENCY; DMGDHD". www.omim.org. Retrieved 2023-12-28.
- ↑ "MYOPATHY WITH MYALGIA, INCREASED SERUM CREATINE KINASE, AND WITH OR WITHOUT EPISODIC RHABDOMYOLYSIS; MMCKR". www.omim.org. Retrieved 2023-12-28.
- ↑ "Glycogen Storage Disease Type I - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Retrieved 23 March 2017.
- ↑ Løkken, Nicoline; Hansen, Kit K.; Storgaard, Jesper H.; Ørngreen, Mette C.; Quinlivan, Ros; Vissing, John (July 2020). "Titrating a modified ketogenic diet for patients with McArdle disease: A pilot study". Journal of Inherited Metabolic Disease. 43 (4): 778–786. doi:10.1002/jimd.12223. ISSN 0141-8955. PMID 32060930. S2CID 211121921.
- ↑ Løkken, Nicoline; Voermans, Nicol C.; Andersen, Linda K.; Karazi, Walaa; Reason, Stacey L.; Zweers, Heidi; Wilms, Gustav; Santalla, Alfredo; Susanibar, Edward; Lucia, Alejandro; Vissing, John (2023-02-07). "Patient-Reported Experiences with a Low-Carbohydrate Ketogenic Diet: An International Survey in Patients with McArdle Disease". Nutrients. 15 (4): 843. doi:10.3390/nu15040843. ISSN 2072-6643. PMC 9964801. PMID 36839201.
- 1 2 Kitaoka, Yu (February 25, 2014). "McArdle Disease and Exercise Physiology". Biology. 3 (1): 157–166. doi:10.3390/biology3010157. ISSN 2079-7737. PMC 4009758. PMID 24833339.
- 1 2 3 4 5 6 Salazar-Martínez, Eduardo; Santalla, Alfredo; Valenzuela, Pedro L.; Nogales-Gadea, Gisela; Pinós, Tomàs; Morán, María; Santos-Lozano, Alejandro; Fiuza-Luces, Carmen; Lucia, Alejandro (2021). "The Second Wind in McArdle Patients: Fitness Matters". Frontiers in Physiology. 12: 744632. doi:10.3389/fphys.2021.744632. ISSN 1664-042X. PMC 8555491. PMID 34721068.
- 1 2 Perez, M; Martin, M A; Rubio, J C; Maté-Muñoz, J L; Gómez-Gallego, F; Foster, C; Andreu, A L; Arenas, J; Lucia, A (August 2006). "Exercise capacity in a 78 year old patient with McArdle's disease: it is never too late to start exercising". British Journal of Sports Medicine. 40 (8): 725–726. doi:10.1136/bjsm.2006.026666. ISSN 0306-3674. PMC 2579473. PMID 16864568.
- ↑ Wakelin, Andrew (2013). 101Tips for a good life with McArdle Disease (PDF). AGSD-UK. p. 52.
- ↑ Pérez, Margarita; Ruiz, Jonatan R.; Fernández del Valle, María; Nogales-Gadea, Gisela; Andreu, Antoni L.; Arenas, Joaquín; Lucía, Alejandro (2009-06-01). "The second wind phenomenon in very young McArdle's patients". Neuromuscular Disorders. 19 (6): 403–405. doi:10.1016/j.nmd.2009.04.010. ISSN 0960-8966. PMID 19477644. S2CID 31541581.
- ↑ Scalco, Renata Siciliani; Morrow, Jasper M.; Booth, Suzanne; Chatfield, Sherryl; Godfrey, Richard; Quinlivan, Ros (September 2017). "Misdiagnosis is an important factor for diagnostic delay in McArdle disease". Neuromuscular Disorders. 27 (9): 852–855. doi:10.1016/j.nmd.2017.04.013. ISSN 1873-2364. PMID 28629675. S2CID 11797963.
- ↑ Stojan, George; Christopher-Stine, Lisa (2015-01-01), Hochberg, Marc C.; Silman, Alan J.; Smolen, Josef S.; Weinblatt, Michael E. (eds.), "151 - Metabolic, drug-induced, and other noninflammatory myopathies", Rheumatology (Sixth Edition), Philadelphia: Mosby, pp. 1255–1263, ISBN 978-0-323-09138-1, retrieved 2023-05-15
- ↑ Applegarth DA, Toone JR, Lowry RB (January 2000). "Incidence of inborn errors of metabolism in British Columbia, 1969–1996". Pediatrics. 105 (1): e10. doi:10.1542/peds.105.1.e10. PMID 10617747. S2CID 30266513.
- ↑ Cantú-Reyna, Consuelo; Zepeda, Luis Manuel; Montemayor, René; Benavides, Santiago; González, Héctor Javier; Vázquez-Cantú, Mercedes; Cruz-Camino, Héctor (27 September 2016). "Incidence of Inborn Errors of Metabolism by Expanded Newborn Screening in a Mexican Hospital" (PDF). Journal of Inborn Errors of Metabolism and Screening. 4: 232640981666902. doi:10.1177/2326409816669027.
External links
- IamGSD - International Association for Muscle Glycogen Storage Disease. A non-profit, patient-led international group encouraging efforts by research and medical professionals, national support groups and individual patients worldwide.
- IPA - International Pompe Association. (Pompe Disease is also known as GSD-II). A non-profit, federation of Pompe disease patient's groups world-wide. It seeks to coordinate activities and share experience and knowledge between different groups.
- EUROMAC - EUROMAC is a European registry of patients affected by McArdle Disease and other rare neuromuscular glycogenoses.
- CoRDS - Coordination of Rare Diseases at Sanford (CoRDS) is a centralized international patient registry for all rare diseases. They work with patient advocacy groups, including IamGSD, individuals and researchers.
- CORD - Canadian Organization for Rare Disorders (CORD) is a Canadian national network for organizations representing all those with rare disorders. CORD provides a strong common voice to advocate for health policy and a healthcare system that works for those with rare disorders.
- NORD - National Organization for Rare Disorders (NORD) is an American national non-profit patient advocacy organization that is dedicated to individuals with rare diseases and the organizations that serve them.
- EURODIS - Rare Diseases Europe (EURODIS) is a unique, non-profit alliance of over 700 rare disease patient organizations across Europe that work together to improve the lives of the 30 million people living with a rare disease in Europe.