苯
| 苯 | |||
|---|---|---|---|
 | |||
| |||
| |||
| IUPAC名 Benzene[1] | |||
| 别名 | 囷、焑、㷍、菕、輪質 1,3,5-環己三烯(理論共振異構體) [6]輪烯(不推薦[1]) | ||
| 识别 | |||
| CAS号 | 71-43-2 | ||
| PubChem | 241 | ||
| ChemSpider | 236 | ||
| SMILES |
| ||
| InChI |
| ||
| InChIKey | UHOVQNZJYSORNB-UHFFFAOYAH | ||
| ChEBI | 16716 | ||
| RTECS | CY1400000 | ||
| 性质 | |||
| 化学式 | C6H6 | ||
| 摩尔质量 | 78.11 g·mol−1 | ||
| 外观 | 无色透明易挥发液体 | ||
| 密度 | 0.8765(20) g/cm³[2] | ||
| 熔点 | 5.5 °C(279 K) | ||
| 沸点 | 78.3 °C(351 K) | ||
| 溶解性(水) | 1.8 g/L(15 ℃)[3][4][5] | ||
| 黏度 | 0.652 cP,20℃时 | ||
| 偶极矩 | 0 D | ||
| 热力学 | |||
| S⦵298K | 173.26 J/mol·K | ||
| 热容 | 135.69 J/mol·K(298.15 K) | ||
| 危险性 | |||
| 警示术语 | R:R45-R46-R11-R36/38-R48/23/24/25-R65 | ||
| 安全术语 | S:S53-S45 | ||
| 欧盟分类 | 可燃(F) Carc. Cat. 1 Muta. Cat. 2 有毒(T) | ||
| NFPA 704 |
 3
3
0
| ||
| 闪点 | −10.11℃(闭杯) | ||
| 自燃温度 | 562.22℃ | ||
| 相关物质 | |||
| 相关化学品 | 甲苯 环硼氮烷 | ||
| 若非注明,所有数据均出自标准状态(25 ℃,100 kPa)下。 | |||
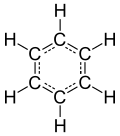
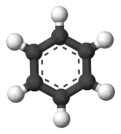


苯是最简单的芳香烃,难溶于水,易溶于有机溶剂,本身也可作有机溶剂。苯是石油化工基本原料,其产量和生产的技术是国家石油化工发展的标志。苯的环系叫苯环,是最简单的芳香环。苯分子去掉一粒氢后的结构叫苯基,用Ph表示;因此苯也可表示为PhH。
发现
苯最早在18世纪初研究将煤气作为照明用气时合成出来。一般认为苯是由麥可·法拉第在1825年发现。他从鱼油等类似物质的热裂解产品中分离出了纯度甚高的苯,称之为“氢的重碳化物”(Bicarburet of hydrogen)。[7]并且测定了苯的一些物理性质和其化学组成,阐述了苯分子的碳氢比。[8]
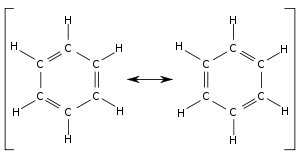
1833年,米修里希·伊尔哈得确定了苯分子中6碳和6氢原子的经验式(C6H6)。[9]1865年,弗里德里希·凯库勒提出了苯环单、双键交替排列、无限共轭的结构(⌬),即现在所谓“凯库勒式”,据称他梦到一条蛇咬住了自己的尾巴才受到启发想出。[10]他解释称环中双键位置不固定,可迅速移动,所以6粒碳等价。他通过对苯的一氯代物、二氯代物种类的研究,发现苯是环形结构,每粒碳连接一粒氢。也有说法指出,把苯分子结构画成六角形环状最早是法国化学家奥古斯特·劳伦1854年在《化学方法》一书中提出。但是出于某种原因,凯库勒在论文没有提及劳伦的成果。[10]
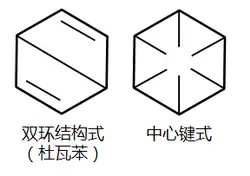
也有人提出了其他设想:比如詹姆斯·杜瓦就歸納出不同结构,以其命名的杜瓦苯现已证实是與苯不同的另一种物质,可由苯经光照得到。
1845年德国化学家霍夫曼从煤焦油的轻馏分中发现了苯,其学生查尔斯·曼斯菲尔德(Charles Mansfield)随后加工提纯了苯。后来他又发明了结晶法精制苯。他还研究工业用途,开创了苯的加工利用途径。大约从1865年起开始工业生产。最初是从煤焦油中回收。随着它用途增多,产量不断上升,到1930年已经成为世界十大吨位产品之一。
结构
苯的苯环结构給它特殊的芳香性。苯环是最简单的芳环,由六粒碳原子构成六元环,每粒碳原子接一組基团,苯的6組基团都是氢原子。
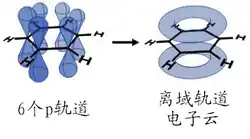
苯不是单、双键交替排列的轮烯,原子间成键并不是不连续的单双键交替,而是給离域π电子云覆盖。
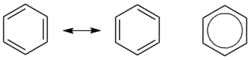
苯分子是平面分子,12粒原子处于同一平面,6粒碳和6粒氢均等,碳-氫键长1.08Å,碳-碳键长1.40Å,此数值介于单双键长之间。分子所有键角均为120°,说明碳原子都采取sp2杂化。这样每粒碳原子还剩余一條p轨道垂直于分子平面,每條轨道有一粒电子。于是6條轨道重叠形成离域大Π键(即π66),有下图所示的共振式,现在认为这是苯环非常稳定的原因,也直接使苯环有芳香性。
从分子轨道理论来看,可以认为苯的6條p轨道相互作用形成6條π分子轨道,其中ψ1、ψ2、ψ3是能量较低的成键轨道,ψ4、ψ5、ψ6是能量较高的反键轨道。ψ2、ψ3和ψ4、ψ5是两对简并轨道。基态时苯的电子云分布是三个成键轨道叠加的结果,故电子雲均匀分布于苯环上下及环原子上,形成闭合的电子雲。它是苯分子在磁场中产生环电流的根源。
物理性质
苯的沸点80.1℃,熔点5.5℃,在常温无色,透明,有芳香气味,易挥发。苯比水密度低,密度为每毫升0.88克,但其分子質量比水高。苯难溶于水,1升水最多溶解1.8克苯;但苯是良好的有机溶剂,溶解有机分子和一些非极性无机分子的能力很强。
苯能与水恒沸,沸点69.25℃,含苯91.2%,在有水生成的反应中常加苯蒸馏,以将水餾出。

P单位为mmHg,t单位为℃,A=6.91210,B=1214.645,C=221.205。
化学性质
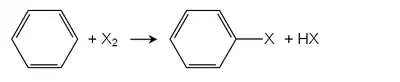
苯可参与的化学反应大致有3种:其他基团和苯环的氢原子间发生取代反应;加成到碳=碳键;苯环断裂。
取代反应
卤素、硝基、磺酸基、烃基等可在一定条件下取代苯环的氢原子,生成相应衍生物。取代基不同以及氢原子位置、数量不同,可以生成不同数量和结构的同分异构体。
苯环电子云密度甚大,在苯环的取代反应大都是亲电取代反应。亲电取代反应是芳环的代表反应。苯的取代物在亲电取代时,第二取代基的位置与原先取代基的种类有关。


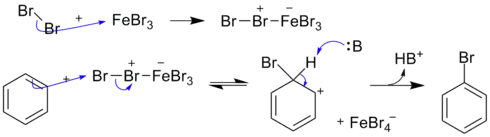
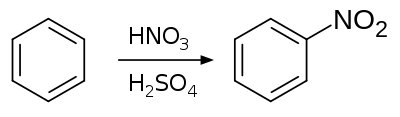
磺化反应

磺化是可逆反应,苯磺酸加热与稀硫酸或盐酸反应,可失去磺基,生成苯。合成时可以先磺化以保护芳核的某位置。
苯环引入磺酸基后反应能力下降,不易繼續磺化,需要更高温才能引入第二、第三組磺酸基。这说明硝基、磺酸基都是钝化基团,即妨碍再次亲电取代的基团。
傅-克反应
三氯化鋁催化,苯和醇、烯烃和卤代烃反应,烷基取代苯环的氢原子生成烷基苯。这反应称为(傅-克)烷基化反应。[11]例如与乙烯烷基化生成乙苯:

反应过程中,烴基可能会重排:如1-氯丙烷与苯反应生成异丙苯,自由基总是趋向稳定的构型。



苯环的亲电取代定位效应
制备
苯可以由含碳量高的物质不完全燃烧获得。自然界中,火山爆发和森林火災都能生成苯。香烟的烟也有苯。
直至二战,苯还是一种钢铁工业焦化过程中的副产物。这种方法只能从1吨煤中提取出1千克苯。1950年代后,随着工业,尤其是日益发展的塑料工业对苯的需求增多,由石油生产苯的过程应运而生。现在全球大部分的苯来源于石油化工。工业上生产苯最重要的三种过程是催化重整、甲苯加氢脱烷基化和蒸汽裂化。
从煤焦油中提取
在煤炼焦过程中生成的轻焦油含有大量的苯。这是最初生产苯的方法。将生成的煤焦油和煤气一起通过洗涤和吸收设备,用高沸点的煤焦油作为洗涤和吸收剂回收煤气中的煤焦油,蒸馏后得到粗苯和其他高沸点馏分。粗苯经过精制可得到工业级苯。这种方法得到的苯纯度比较低,而且环境污染严重,工艺比较落后。
从石油中提取
原油中含有少量的苯,从石油产品中提取苯是最广泛使用的制备方法。
催化重整
重整这里指使脂肪烃成环、脱氢形成芳香烃的过程。这是从第二次世界大战期间发展形成的工艺。
在500至525 ℃、8至50大气压下,各种沸点在60至200℃之间的脂肪烃,经铂-铼催化剂,通过脱氢、环化转化为苯和其他芳香烃。从混合物中萃取出芳香烃产物后,再经蒸馏即分出苯。也可以将这些馏分用作汽油。
蒸汽裂解
蒸汽裂解是由乙烷、丙烷或丁烷等低分子烷烃以及石脑油、重柴油等石油组份生产烯烃的一种过程。其副产物之一裂解汽油富含苯,可以分馏出苯及其他各种成分。裂解汽油也可与其他烃类混合作为汽油添加剂。
裂解汽油大约有40至60%苯,同时还含有二烯烃以及苯乙烯等其他不饱和组份,这些杂质在贮存过程中易进一步反应生成高分子胶质。所以要先经过加氢处理过程来除去裂解汽油中的这些杂质和硫化物,然后再适当地分离得到苯产品。
芳烃分离
从不同方法得到的含苯馏分,组分非常复杂,用普通的分离方法很难见效,一般用溶剂液-液萃取或者萃取蒸馏的方法分离芳烃,然后再采用一般的分离方法分离苯、甲苯、二甲苯。根据采用的溶剂和技术的不同又有多种分离方法。
- Udex法:由美国道化学公司和UOP公司在1950年联合开发,最初用二乙二醇醚作溶剂,后来改进为三乙二醇醚和四乙二醇醚作溶剂,过程采用多段升液通道(multouocomer)萃取器。苯的收率为100%。
- Suifolane法:荷兰壳牌公司开发,专利为UOP公司所有。溶剂采用环丁砜,使用转盘萃取塔萃取,产品需经白土处理。苯的收率为99.9%。
- Arosolvan法:由联邦德国的鲁奇公司在1962年开发。溶剂为N-甲基吡咯烷酮(NMP),为了提高收率,有时还加入10至20%乙二醇醚。采用特殊设计的Mechnes萃取器,苯的收率为99.9%。
- IFP法:由法国石油化学研究院在1967年开发。采用不含水的二甲亚砜作溶剂,并用丁烷反萃取,过程采用转盘塔。苯的收率为99.9%。
- Formex法:为意大利SNAM公司和LRSR石油加工部在1971年开发。吗啉或N-甲酰吗啉作溶剂,采用转盘塔。芳烃总收率98.8%,其中苯的收率为100%。
甲苯脱烷基化
甲苯脱烷基制备苯,可以采用催化加氢脱烷基化,或是不用催化剂的热脱烷基。原料可以用甲苯、及其和二甲苯的混合物,或者含有苯及其他烷基芳烃和非芳烃的馏分。
甲苯催化加氢脱烷基化
用铬、钼或氧化铂等作催化剂,500至600℃高温和400至600大气压,甲苯与氢气混合可以生成苯,过程称为加氢脱烷基化作用。如果温度更高,则可以省去催化剂。按照以下方程式反应:

根据所用催化剂和工艺条件的不同又有多种工艺方法:
- Hydeal法:由Ashiand & refing和UOP公司在1961年开发。原料可以是重整油、加氢裂解汽油、甲苯、6碳至8碳混合芳烃、脱烷基煤焦油等。催化剂为氧化铝-氧化铬,反应温度600至650℃,压力3.43至3.92MPa。苯的理论收率为98%,纯度可达99.98%以上,质量优于Udex法生产的苯。
- Detol法:Houdry公司开发。用氧化铝和氧化镁做催化剂,反应温度540至650℃,反应压力0.69至5.4MPa,原料主要是7碳至9碳芳烃。苯的理论收率为97%,纯度可达99.97%。
- Pyrotol法:Air products and chemicals公司和Houdry公司开发。适用于从乙烯副产裂解汽油中制苯。催化剂为氧化铝-氧化铬,反应温度600至650℃,压力0.49至5.4MPa。
- Bextol法:壳牌公司开发。
- BASF法:BASF公司开发。
- Unidak法:UOP公司开发。
甲苯热脱烷基
甲苯在高温氢气流中可不催化就脱烷基生成苯。反应为放热反应,针对遇到的不同问题,开发出了多种工艺过程。
- MHC加氢脱烷基过程:由日本三菱石油化学公司和千代田建设公司在1967年开发。原料可以用甲苯等纯烷基苯,含非芳烃30%以内的芳烃馏分。操作温度500至800℃,操作压力0.98MPa,氢/烃比为1至10。过程选择性97至99%(摩比),产品纯度99.99%。
- HDA加氢脱烷基过程:由美国Hydrocarbon Research和Atlantic Richfield公司在1962年开发。原料采用甲苯,二甲苯,加氢裂解汽油,重整油。从反应器不同部位同如氢气控制反应温度,反应温度600至760℃,压力3.43至6.85MPa,氢/烃比为1至5,停留时间5至30秒。选择性95%,收率96至100%。
- Sun过程:由Sun Oil公司开发
- THD过程:Gulf Research and Development公司开发
- Monsanto过程:孟山都公司开发
甲苯歧化和烷基转移
二甲苯用量上升,在1960年代末相继开发出可以同时增产二甲苯的甲苯歧化和烷基转移技术,主要反应为:
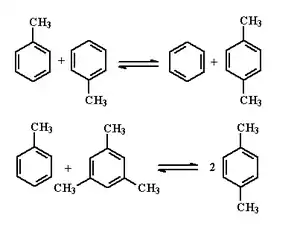
这反应可逆,工艺过程因催化剂、工艺条件、原料而異。
- LTD液相甲苯歧化过程:美国美孚化学公司在1971年开发,使用非金属沸石或分子筛催化剂,反应温度260至315℃,反应器采用液相绝热固定床,原料为甲苯,转化率99%以上
- Tatoray过程:日本东丽公司和UOP公司1969年开发,以甲苯和混合碳9芳烃为原料,催化剂为丝光沸石,[14]反应温度350至530℃,压力2.94MPa,氢/烃比5至12,采用绝热固定床反应器,单程转化率40%以上,收率95%以上,选择性90%,产品为苯和二甲苯混合物。
- Xylene plas过程:由美国Atlantic Richfield公司和Engelhard公司开发。使用稀土Y型分子筛做催化剂,反应器为气相移动床,反应温度471至491℃,常压。
- TOLD过程:日本三菱瓦斯化学公司1968年开发,氢氟酸-氟化硼催化剂,反应温度60至120℃,低压液相。有一定腐蚀力。

分析测试方法
气相色谱和液相色谱可以检测各种产品的苯含量。苯的纯度一般用冰点法测定。
检测空气中的微量苯,可以用甲基硅油等易挥发有机溶剂或者低分子量聚合物吸收,然后用色谱分析;或者采用比色法分析;也可以将含有苯的空气深度冷冻,将苯冷冻下来,然后把硫酸铁和过氧化氢溶液加入得到黄褐色或黑色沉淀,再用硝酸溶解,然后用比色法分析。或者直接用硝酸吸收空气中的苯,硝化成间二硝基苯,然后用二氯化钛溶液滴定,或者用间二甲苯配制的丁酮碱溶液比色定量。
安全
有毒
苯易挥发,暴露于空气中很易扩散。人和动物吸入或皮肤接触大量苯进入体内,会引起急性和慢性苯中毒。有研究报告表明,引起苯中毒的部分原因是苯在体内转化为苯酚。
苯会麻痹中枢神经系统,引起急性中毒。重者会出现头痛、恶心、呕吐、神志模糊、知觉丧失、昏迷、抽搐等症状,严重者会因中枢系统麻痹而死亡。少量苯也能使人产生睡意、头昏、心率加快、头痛、颤抖、意识混乱、神志不清等现象。摄入含苯过多的食物会引起呕吐、胃痛、头昏、失眠、抽搐、心率加快等症状,甚至死亡。吸入20000ppm的苯蒸气5至10分钟便会有致命危险。
长期接触苯会对血液造成极大伤害,引起慢性中毒,引起神经衰弱综合症。苯可以损害骨髓,使红血球、白细胞、血小板数量减少,并使染色体畸变,从而导致白血病,甚至出现再生障碍性贫血。苯可以导致大量出血,从而抑制免疫系统的功用,使疾病有机可乘。有研究报告指出,苯在体内的潜伏期可长达12至15年。
妇女吸入过量苯后,会导致月经不调达数月,卵巢会缩小。苯对胎儿发育和对男性生殖力的影响尚未明了。孕期动物吸入苯后,会导致幼体的重量不足、骨骼延迟发育、骨髓损害。
苯对皮肤、粘膜有刺激作用。国际癌症研究中心(IARC)已经确认苯为致癌物。[15][16]
接触限值:
毒性:
- LD50:930 mg/kg(大鼠经口);48 mg/kg(小鼠经皮)
- LC50:10000ppm 7小时(大鼠吸入)
当然,每个人的健康状况和接触条件不同,对苯的敏感程度也不相同。嗅出苯的气味时,它的浓度大概是1.5ppm,这时就应该警惕中毒的危险。检查时,尿和血液可以很易查出苯的中毒程度。
代谢
苯主要經呼吸道吸入(47至80%)、胃肠及皮肤吸收的方式进入人体。一部分苯可由尿液排出,未排出的苯则首先在肝中细胞色素P450单加氧酶作用下給氧分子氧化为环氧苯(7-氧杂双环[4.1.0]庚-2,4-二烯)。环氧苯与它的重排产物氧杂环庚三烯存在平衡,是苯代谢过程中产生的有毒中间体。接下来有三种代谢途径:与谷胱甘肽结合生成苯巯基尿酸;继续代谢为苯酚、邻苯二酚、对苯二酚、偏苯三酚、邻苯醌、对苯醌等,以葡萄糖苷酸或硫酸盐结合物形式排出;以及氧化为已二烯二酸。
可燃
苯可在空气中燃烧,一般都定为危险品。例如在中华人民共和国国家标准《危险货物品名表》(GB 12268-90)中,苯属第三类危险货物易燃液体中的中闪点液体。[17]而且它甚易挥发,可能造成蒸气局部聚集,贮存、运输时一般都要求远离火源和热源,防止静电。
苯的冰点頗高(5.5℃),在寒冷天气中运输会有困难,但是加热熔化会有危险。
工业用途
早在1920年代,苯就已是工业常用的溶剂,主要用于金属脱脂。苯有毒,人体能直接接触溶剂的生产过程现已不用苯作溶剂。
苯有减轻爆震的作用而能作为汽油添加剂。在1950年代四乙基铅开始使用以前,所有抗爆剂都是苯。然而现在含铅汽油淡出,苯又重新起用。苯对人体有不利影响,对地下水质也有污染,欧美国家限定汽油中苯的含量不得超过1%。2011年,美國國家環境保護局再次收緊限制,汽油的苯含量上限降至0.62%。[18]
苯在工业上最重要的用途是做化工原料。苯可以合成一系列苯的衍生物:
食品中的含量
在若干軟性飲品,如部份汽水、果汁飲品、果汁味飲品、葡萄適中,苯甲酸盐类防腐剂(包括苯甲酸鈉、苯甲酸钾、苯甲酸钙)[19]会跟維他命C(抗坏血酸)或异抗坏血酸发生化學作用,脱羧,形成少量致癌物質苯,光照或加热时加速反应。多数饮料中,苯的含量都在每公斤10μg以下,符合世界卫生组织标准(10ppb),但高于美国(5ppb)[20]、加拿大(5ppb)和欧盟(1ppb)[21]的标准。有少量饮料含有较高含量的苯,含量最高的可达87.9ppb,但与日常生活中吸入的苯含量相比,这个数值仍然较小。例如,人每天吸入的洁净空气中约含苯220μg;人开车一小时会吸入40μg苯;每天吸20支烟的人约会吸入7900μg苯(欧盟估计值),經二手烟吸入的苯也有63μg。[22]
苯的衍生物
下面是一些苯的著名取代物或与苯结构相似的物质。
參见
- 芳香性
- 粗苯
参考文献
引用
- Favre, Henri A.; Powell, Warren H. . Cambridge: The Royal Society of Chemistry. 2014: 10, 22, 204, 494, 577. ISBN 978-0-85404-182-4. doi:10.1039/9781849733069.
- David R. Lide, ed. . CRC Press.
- Arnold, D.; Plank, C.; Erickson, E.; Pike, F. . Industrial & Engineering Chemistry Chemical & Engineering Data Series. 1958, 3: 253. doi:10.1021/i460004a016.
- Breslow, R; Guo, T. . Proceedings of the National Academy of Sciences of the United States of America. 1990, 87 (1): 167–9. PMC 53221
 . PMID 2153285. doi:10.1073/pnas.87.1.167.
. PMID 2153285. doi:10.1073/pnas.87.1.167. - A. Kayode Coker, Ernest E. Ludwig. . Elsevier. 2007: 114 [2010-09-13]. ISBN 075067766X. (原始内容存档于2020-11-15).
- . [2019-01-24]. (原始内容存档于2021-05-10).
- M. Faraday. . Philosophical Transactions of the Royal Society of London. 1825, 115: 440–466. doi:10.1098/rstl.1825.0022.
- R. Kaiser. . Angewandte Chemie International Edition in English. 1968, 7 (5): 345–350. doi:10.1002/anie.196803451.
- E. Mitscherlich. . Annalen der Pharmacie. 1834, 9 (1): 39–48. doi:10.1002/jlac.18340090103.
- . [2009-01-26]. (原始内容存档于2020-11-07).
- Friedel, C.; Crafts, J. M. Compt. Rend. 1877, 84, 1392 (页面存档备份,存于) & 1450 (页面存档备份,存于).
- Biphenyl(1,1- Biphenyl). Wiley/VCH, Weinheim (1991), ISBN 978-3-527-28277-7
- 曾昭琼。有机化学.ISBN 978-7-04-013845-0
- 謝在庫,陳慶齡,張成芳. . 化工進展. 1999, 第2期 [2009-02-20]. (原始内容存档于2016-03-12).
- Huff J. . Int J Occup Environ Health. Apr–Jun 2007, 13 (2): 213–21. PMID 17718179.
- Rana SV; Verma Y. . J Environ Biol. April 2005, 26 (2): 157–68. PMID 16161967.
- 中闪点液体列表 (页面存档备份,存于) - 中华人民共和国《危险货物品名表》(GB 12268-90)
- . U.S. Environmental Protection Agency: 15853. 2006-03-29 [2008-06-27]. (原始内容存档于2008-12-05).
- LK Gardner, GD Lawrence, Benzene Production from Decarboxylation of Benzoic Acid in the Presence of Ascorbic Acid and a Transition-Metal Catalyst, Journal of Agricultural and Food Chemistry, May 1993, Volume 41, Number 5; first page (页面存档备份,存于)
- United States Food and Drug Administration: Questions and Answers on the Occurrence of Benzene in Soft Drinks and Other Beverages 的存檔,存档日期2008-03-26.
- Council Directive 98/83/EC of 3 November 1998 on the quality of water intended for human consumption Archived 2010-09-17 at WebCite(PDF)
- New Zealand Food Safety Authority / Te Pou Oranga kai O Aotearoa Benzene in flavoured drinks 的存檔,存档日期2007-04-23.
- Martin Hickman Coca-Cola to phase out use of controversial additive after DNA damage claim 的存檔,存档日期2010-04-23. The Independent 25 May 2008
- [. [2011-12-29]. (原始内容存档于2021-05-10). Benzene字典-Guidechem.com(英文)]
书籍
- 魏文德主编,《有机化工原料大全》第三卷,化学工业出版社,1994年,p358至381,ISBN 978-7-5025-0684-1
- [英]汉考克(Hancock, E.G.)主编,《苯及其工业衍生物》,化学工业出版社,1982.11
- Wilson, L. D. "Health Hazards from aromatic Hydrocarbons", Des Plaines, III., Universal Oil Products Company, 1962
- 尹冬冬主编,《有机化学》上册,高等教育出版社,2003年,ISBN 978-7-04-011055-5
其他
- 中国石化北京化工研究院,《常用危险化学品安全数据卡》(内部材料),2004年
外部链接
| 维基词典中的词条「」。 |
| 维基共享资源中相关的多媒体资源:苯 |
- Benzene Material Safety Data Sheet
- Chemistry WebBook的化学性质数据 (页面存档备份,存于)
- 职业性苯中毒诊断标准——GBZ68-2002
- 化工世界苯网——提供苯的市场行情