G蛋白偶联受体
G蛋白偶联受体(G protein-coupled receptor,GPCRs),是一大类膜蛋白受体的统称。
| GPCR | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 | |||||||||
| 鑑定 | |||||||||
| 標誌 | 7tm_1 | ||||||||
| Pfam | PF00001(旧版) | ||||||||
| InterPro | IPR000276 | ||||||||
| PROSITE | PDOC00210 | ||||||||
| OPM家族 | 6 | ||||||||
| OPM蛋白 | 1gzm | ||||||||
| |||||||||
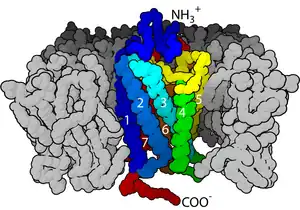
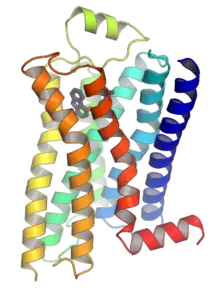
这类受体的共同点是其立体结构中都有七个跨膜α螺旋,且其肽链的C端和连接第5和第6个跨膜螺旋的胞内环上都有G蛋白(鸟苷酸结合蛋白)的结合位点。
目前为止,只在真核生物中发现G蛋白偶联受体。它们参与很多细胞信号转导过程。在这些过程中,G蛋白偶联受体能结合细胞周围环境中的化学物质并激活细胞内的一系列信号通路,最终引起细胞状态的改变。已知的与G蛋白偶联受体结合的配体包括气味分子、费洛蒙、荷尔蒙、神经递质、趋化因子等等。这些受体可以是小分子的糖类、脂质、多肽,也可以是蛋白质等生物大分子。一些特殊的G蛋白偶联受体也可以被非化学性的刺激源激活,例如在感光细胞中的视紫红质可以被光所激活。与G蛋白偶联受体相关的疾病为数众多,并且大约40%的现代药物都以G蛋白偶联受体作为靶点。[2][3]
G蛋白偶联受体的下游信号通路有多种。与配体结合的G蛋白偶联受体会发生构象变化,从而表现出鸟苷酸交换因子(GEF)的特性,通过以三磷酸鸟苷(GTP)交换G蛋白上本来结合着的二磷酸鳥苷(GDP)使G蛋白的α亚基与β、γ亚基分离。这一过程使得G蛋白(特别地,指其与GTP结合着的α亚基)变为激活状态,并参与下一步的信号传递过程。具体的传递通路取决于α亚基的种类(Gαs、Gαi/o、Gαq/11、Gα12/13)。[4]:1160其中主要的两个通路分别以由三磷酸腺苷环化产生的环腺苷酸(cAMP)和由磷脂酰肌醇-4,5-二磷酸(PIP2)水解生成的肌醇三磷酸(IP3)和甘油二酯(DAG)作为第二信使,[5] 详见环腺苷酸信号通路和磷脂酰肌醇信号通路。
历史与重要性
2011年确定了第一个G蛋白偶联受体(GPCR)与G蛋白三聚体(Gαβγ)复合体的结构,为GPCR研究开辟了一个新的篇章,使得涉及多个蛋白质的全局开关的结构研究成为可能。此前的重大突破包括在2000年确定了第一个GPCR,视紫红质的晶体结构,以及在2007年确定了第一个具有可扩散配体的GPCR(β2AR)的晶体结构。基于对两维晶体冷冻电子显微镜研究的低分辨率模型,人们猜测了GPCR的七个跨膜螺旋如何排列成一个束。三年后出现的视紫红质晶体结构除了揭示了一个额外的细胞质螺旋H8和精确定位视黄醛结合位点的环之外,并没有太多意外。然而,它提供了一个骨架,希望能成为其他GPCR的同源建模和药物设计的通用模板——这种观念被证明过于乐观。
七年后,带有可扩散配体的β2-肾上腺素受体(β2AR)的结晶揭示了出乎意料的结果,因为它展示了与视紫红质不同的受体胞外侧形状。这一区域非常重要,因为它负责配体结合,并且是许多药物的靶点。此外,与视紫红质结构相比,配体结合位点更加宽敞,并向外部开放。随后短时间内结晶的其他受体中,结合位点对配体的可接触性甚至更强。新的结构与生化研究揭示了分子开关的作用机制,这些开关调节受体的结构,导致激动剂的激活状态或逆激动剂的完全或部分失活状态。
2012年的诺贝尔化学奖授予了Brian Kobilka和Robert Lefkowitz,以表彰他们在“理解G蛋白偶联受体功能方面的关键工作”。[6] 至少还有其他七项诺贝尔奖授予了与G蛋白介导的信号传导相关的某些方面的研究。截至2012年,全球十大畅销药物中至少有两种(Advair Diskus和Abilify)是通过靶向G蛋白偶联受体而发挥作用。[7]
分类
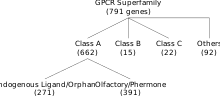
G蛋白偶联受体组成了一个非常庞大的蛋白质超家族(protein superfamily)。这个超家族的具体大小尚未知晓,不过依据DNA序列的相似性,人们预测人类基因组中有约800个基因(约占整个基因组中编码蛋白质的部分的4%)会编码G蛋白偶联受体超家族的成员蛋白。[9][8] 这些G蛋白偶联受体可以被大致划分为六个类型,分属其中的G蛋白偶联受体的基因序列之间没有显著的同源关系。[10][11][12][13]
- A 类(或 第一类)(视紫红质样受体)
- B 类(或 第二类)(分泌素受体家族)
- C 类(或 第三类)(代谢型谷氨酸受体/信息素)
- D 类(或 第四类)(真菌交配信息素受体)
- E 类(或 第五类)(环腺苷酸受体)
- F 类(或 第六类)(卷曲受体(Frizzled)/Smoothened家族)
其中第一类即视紫红质样受体包含了绝大多数种类的G蛋白偶联受体。它被进一步分为了19个子类A1-A19。[14]最近,有人提出了一种新的关于G蛋白偶联受体的分类系统,被称为GRAFS,即谷氨酸(Glutamate),视紫红质(Rhodopsin),粘附(Adhesion),Frizzled/Taste2以及分泌素(Secretin)的英文首字母缩写。[15]
一些基于生物信息学的研究着眼于预测那些具体功能尚未明了的G蛋白偶联受体的分类。[16][17][18]研究者使用被称为伪氨基酸组成的方法利用G蛋白偶联受体的氨基酸系列来预测它们在生物体内可能的功能以及分类。
受体结构
G蛋白偶联受体均是膜内在蛋白(Integral membrane protein),每个受体内包含七个α螺旋组成的跨膜结构域,这些结构域将受体分割为膜外N端(N-terminus),膜内C端(C-terminus),3个膜外环(Loop)和3个膜内环。受体的膜外部分经常带有糖基化修饰。膜外环上包含有两个高度保守的半胱氨酸残基,它们可以通过形成二硫键稳定受体的空间结构。有些光敏感通道蛋白(Channelrhodopsin)和G蛋白偶联受体有着相似的结构,也包含有七个跨膜螺旋,但同时也包含有一个跨膜的通道可供离子通过。
与G蛋白偶联受体相似,PAQR家族蛋白(包括两种脂联素受体ADIPOR1和2)也包含七个跨膜域,但是它们以相反的方向跨于膜上(即N端在膜内而C端在膜外),并且它们也不与G蛋白相互作用。[19]
早期关于G蛋白偶联受体结构的模型是基于他们与细菌视紫红质(Bacteriorhodopsin)之间微弱的趋同演化关系的,其中后者的结构已由电子衍射(蛋白质数据库资料编号:PDB 2BRD和PDB 1AT9)[20][21]和X射线晶体衍射(PDB 1AP9)实验所获得。[22]在2000年,第一个哺乳动物G蛋白偶联受体——牛视紫红质的晶体结构(PDB 1F88)被解出。[23] 2007年,第一个人类G蛋白偶联受体的结构(PDB 2R4R和PDB 2R4S)被解出。[24]随后不久,同一个受体的更高解析度的结构(PDB 2RH1)被发表出来。[1][25]这个人G蛋白偶联受体——β2肾上腺素能受体,显示出与牛视紫红质的高度相似,不过两者在第二个膜外环的构象上完全不同。由于第二膜外环组成了一个类似盖子的结构罩住了配体结合位点,这个构象上的区别使得所有对从视紫红质建立G蛋白偶联受体同源结构模型的努力变得困难重重。
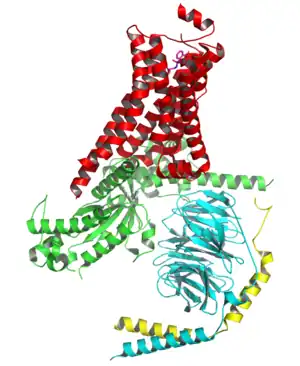
一些激活的即结合了配体的G蛋白偶联受体的结构也已经被研究清楚。[26][27][28][29]这些结构显示了G蛋白偶联受体的膜外部分与配体结合了之后会导致膜内部分发生构象变化。其中最显著的变化是第五和第六跨膜螺旋之间的膜内环会向外移动,而激活的β2肾上腺素能受体与G蛋白形成的复合体的结构显示了G蛋白α亚基正是结合在了上述运动所产生的一个空穴处。[30]
生理作用
G蛋白偶联受体参与众多生理过程。包括但不限于以下例子:
- 感光:视紫红质是一大类可以感光的G蛋白偶联受体。它们可以将电磁辐射信号转化成细胞内的化学信号,引导这一过程的反应称为光致异构化(Photoisomerization)。具体细节为:由视蛋白(Opsin)和辅因子视黄醛共价连接所构成的视紫红质在光源的刺激下,分子内的视黄醛会发生异构化,从“11-顺式”变成“全反式”,这个变化进一步引起视蛋白的构象变化从而激活与之偶联的G蛋白,引发下游的信号传递过程。[31][32][33]
- 味觉感觉(味道):味细胞中的GPCR响应苦味和甜味物质介导味觉素(Gustducin)的释放。
- 嗅觉:鼻腔内的嗅上皮(Olfactory epithelium)和犁鼻器上分布有很多嗅觉受体,可以感知气味分子和费洛蒙。
- 行为和情绪的调节:哺乳动物的脑内有很多掌控行为和情绪的神经递质对应的受体是G蛋白偶联受体,包括血清素,多巴胺,γ-氨基丁酸和谷氨酸等。
- 免疫系统的调节:很多趋化因子通过G蛋白偶联受体发挥作用,这些受体被统称为趋化因子受体。其它属于此类的G蛋白偶联受体包括白介素受体(Interleukin receptor)和参与炎症与过敏反应的组胺受体(Histamine receptor)等。
- 自主神经系统的调节:在脊椎动物中,交感神经和副交感神经的活动都受到G蛋白偶联受体信号通路的调节,它们控制着很多自律的生理功能,包括血压,心跳,消化等。
- 细胞密度的调节:最近在盘基网柄菌中发现了一种含有脂质激酶活性的G蛋白偶联受体,可以调控该种黏菌对细胞密度的感应。[34]
- 维持稳态:例如机体内水平衡的调节。[35]
- 参与某些类型肿瘤的生长和遠端轉移[36]。
机理
G蛋白偶联受体传递信号的机理包括几个主要步骤:首先来自细胞膜外侧的配体与受体相结合,引起后者的构象变化,这个过程也称为受体的激活。发生了构象变化的受体随即会激活附着在其细胞膜内侧端的G蛋白,表现为G蛋白上原先结合的GDP被替换为GTP。激活后的G蛋白会进一步引发一系列的下游效应,其中所涉及的具体信号通路则取决于G蛋白的种类。
配体结合位点
大部分A类受体的配体结合部位处于跨膜螺旋和胞外环附近,不过也有一些例外,如糖蛋白激素受体(GPHR)和富亮氨酸重复G蛋白偶联受体(LGR)等。其它类型的G蛋白偶联受体则主要以N端与配体结合。[37] 也有一些报道指出B类受体的跨膜螺旋上也存在潜在的变构配体结合位点。[38]
构象变化
在静息状态下,G蛋白偶联受体在膜上与由Gα、Gβ和Gγ三个亚基组成的异三聚体G蛋白结合形成复合物。其中Gα亚基上结合有GDP分子。当有配体结合到受体上时会引起后者的构象发生变化,变成具有鸟苷酸交换因子活性的“活化构象”。活化的受体会催化Gα亚基捕获GTP分子来交换其上结合着的GDP。GTP与Gα亚基的结合会使受体与G蛋白的复合物解离,受体、GTP-Gα和Gβ-Gγ二聚体三者相互分开。其中后两者可以进一步与其它蛋白相互作用从而使信号继续传递下去,而自由的受体可以重新结合上一个新的G蛋白来开始下一轮信号转导过程。[39]
配体
除了部分情况下可由可见光激活外,生理条件下正常工作的G蛋白偶联受体主要由配体分子所激活。可以与G蛋白偶联受体结合的配体涵盖了很多种类的信号分子,包括各种气味分子、腺苷、肝细胞生长因子(HGF)、一些生物胺(如多巴胺、肾上腺素、去甲肾上腺素、组胺和血清素等)、谷氨酸(通过代谢型谷氨酸受体)、胰高血糖素、乙酰胆碱(通过毒蕈碱型乙酰胆碱受体)、大麻素、趋化因子、介导炎症反应的一些脂质(如前列腺素等类花生酸、血小板活化因子和白三烯等)以及众多肽类激素(如降钙素、生长抑素、生长激素、部分血管活性肠肽家族的成员、抗利尿激素、过敏毒素、促卵泡激素、促性腺激素释放激素、速激肽、缓激肽、蛙皮素、内皮素、γ-氨基丁酸、黑素皮质激素、神经肽Y、阿片肽、促甲状腺素释放激素和催产素等)。另外,也有些G蛋白偶联受体虽然与其它已确认的受体结构上明显相似,但是其內源配体尚未被发现,这样的G蛋白偶联受体同其它类似的受体一起被归类为孤儿受体。
信号传导
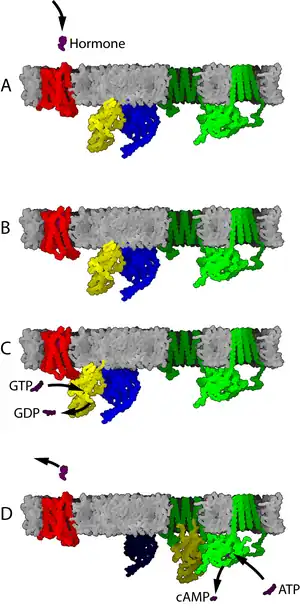
如果一个活跃状态的受体遇到了G蛋白,它可能会激活它。有证据表明受体和G蛋白实际上是预先耦合的。例如,G蛋白与受体的结合会影响受体对配体的亲和力。激活的G蛋白与GTP结合。
后续的信号传导取决于G蛋白的类型。酶腺苷酸环化酶是由G蛋白调节的细胞蛋白的一个例子,在这种情况下是G蛋白Gs。当腺苷酸环化酶与激活的G蛋白的亚单位结合时,其活性被激活。腺苷酸环化酶的激活在G蛋白回到GDP结合状态时结束。
腺苷酸环化酶(人类已知有9种膜结合型和1种细胞质型)也可以通过其他方式(例如Ca2+/钙调蛋白结合)被激活或抑制,这可以与G蛋白一起以加和或协同的方式调节这些酶的活性。
通过GPCR激活的信号通路受限于GPCR本身的一级序列和三级结构,但最终由特定配体稳定的特定构象以及传感器分子的可用性决定。目前,GPCR被认为主要利用两种类型的传感器:G蛋白和β-阿雷斯丁。由于β-阿雷斯丁只对大多数GPCR的磷酸化形式具有高亲和力(见上文或下文),因此大部分信号传导最终依赖于G蛋白的激活。然而,交互作用的可能性确实允许发生G蛋白独立的信号传导。
G蛋白依赖的信号传导
有三个主要的G蛋白介导的信号通路,由四个亚类的G蛋白介导,它们彼此之间通过序列同源性区分开来(Gαs,Gαi/o,Gαq/11,和Gα12/13)。每个亚类的G蛋白由多个基因或剪接变异的产物组成,这些可能赋予它们从微妙到显著的不同,关于信号传导属性,但总的来说,它们似乎合理地分为四个类。因为各种可能的βγ组合的信号传导性质似乎并没有从彼此大不相同,所以这些类别是根据它们α亚单位的同种型来定义的。[4]:1163
虽然大多数GPCR能够激活多个Gα亚型,但它们也表现出对一个亚型的偏好。当激活的亚型取决于与GPCR结合的配体时,这被称为功能选择性(也称为激动剂导向的交通或构象特异性激动作用)。然而,任何单个特定的激动剂的结合也可能启动多个不同的G蛋白,因为它可能能够稳定GPCR的GEF结构域的不止一种构象,甚至在单次相互作用的过程中也是如此。此外,一种优先激活一个Gα同种型的构象可能在首选的Gα同种型较少可用时激活另一个。此外,反馈途径可能导致受体修饰(例如磷酸化),改变G蛋白的偏好。尽管有这些各种细微差别,GPCR的首选耦合伙伴通常根据在大多数生理或实验条件下内源性配体最明显激活的G蛋白来定义。
Gα信号传导
Gαs和Gαi/o途径的效应器是生成环腺苷酸(cAMP)的酶腺苷酸环化酶,或AC。虽然哺乳动物有10种不同的AC基因产物,每种都在组织分布或功能上有细微差别,但所有AC都催化细胞质腺苷三磷酸(ATP)转化为cAMP,并且都直接受到Gαs类G蛋白的刺激。然而,与Gαi/o型的Gα亚单位相互作用则抑制AC生成cAMP。因此,与Gαs耦合的GPCR抵消了与Gαi/o耦合的GPCR的作用,反之亦然。胞浆cAMP水平可能决定了各种离子通道以及丝/苏特异性蛋白激酶A(PKA)家族成员的活性。因此,cAMP被认为是一种第二信使,PKA是次级效应器。
Gαq/11途径的效应器是磷脂酶C-β(PLCβ),它催化膜结合的磷脂酰肌醇4,5-二磷酸(PIP2)分裂成第二信使肌醇(1,4,5)三磷酸(IP3)和二酰甘油(DAG)。IP3作用于内质网(ER)膜上的IP3受体,诱导ER释放Ca2+,而DAG沿着质膜扩散,可能激活膜上的第二丝氨酸/苏氨酸激酶,称为蛋白激酶C(PKC)。由于许多PKC亚型也会因细胞内Ca2+增加而激活,因此这两条途径也可以互相汇合,通过同一个次级效应器进行信号传导。细胞内Ca2+的增加也会结合并异构调节称为钙调蛋白的蛋白质,这些蛋白质反过来又能激活胞质中的小GTP酶,Rho。一旦与GTP结合,Rho就可以激活负责细胞骨架调节的各种蛋白质,如Rho激酶(ROCK)。大多数与Gα12/13耦合的GPCR也与其他亚类耦合,通常是Gαq/11。
Gβγ信号传导
上述描述忽略了Gβγ信号传导的影响,这也可能是重要的,特别是在激活的Gαi/o耦合的GPCR的情况下。Gβγ的主要效应器是各种离子通道,如G蛋白调节的内向整流K+通道(GIRKs)、P/Q型和N型电压门控Ca2+通道,以及某些AC和PLC的亚型,以及一些磷酸肌醇-3-激酶(PI3K)亚型。
G蛋白独立的信号传导
虽然它们通常被认为只是一起工作,但GPCR也可能通过G蛋白独立的机制进行信号传导,而异三聚体G蛋白可能在GPCR独立的功能中发挥作用。GPCR可以通过许多已经提到的在G蛋白依赖的信号传导中发挥作用的蛋白质独立进行信号传导,如β-阿雷斯丁、GRKs和Srcs。这种信号传导已被证明在生理上是相关的,例如,由趋化因子受体CXCR3介导的β-阿雷斯丁信号传导对激活的T细胞的完全有效趋化作用是必需的。[40] 此外,涉及GPCR细胞定位的进一步支架蛋白(例如,含PDZ结构域的蛋白)也可能充当信号转导器。最常见的效应器是MAPK家族成员。
例子
在20世纪90年代末,开始积累的证据表明,一些GPCR能够在没有G蛋白的情况下进行信号传导。ERK2丝裂原激活蛋白激酶,是许多途径中受体激活下游的关键信号传导介质,已被证明在粘菌D. discoideum中对cAMP介导的受体激活做出反应而被激活,尽管缺乏相关的G蛋白α和β亚单位。[41]
在哺乳动物细胞中,广泛研究的β2-肾上腺素受体已被证明在阿雷斯丁介导的G蛋白信号传导解耦后激活ERK2途径。因此,一些先前被认为纯粹与受体脱敏有关的机制实际上可能是受体切换其信号通路的例子,而不仅仅是被关闭。
在肾细胞中,已显示激肽B2受体直接与一种蛋白酪氨酸磷酸酶相互作用。B2受体中存在酪氨酸磷酸化的ITIM(免疫受体酪氨酸基抑制基序)序列是介导这种相互作用及随后激肽的抗增殖作用所必需的。[42]
cAMP和PIP2信号途径的详细信息
涉及G蛋白偶联受体的两个主要信号传导途径是cAMP信号途径和磷脂酰肌醇信号途径。
cAMP信号途径
cAMP信号传导包含五个主要角色:刺激性激素受体(Rs)或抑制性激素受体(Ri);刺激性调节性G蛋白(Gs)或抑制性调节性G蛋白(Gi);腺苷酸环化酶;蛋白激酶A(PKA);以及cAMP磷酸二酯酶。
刺激性激素受体(Rs)是一种能与刺激性信号分子结合的受体,而抑制性激素受体(Ri)是一种能与抑制性信号分子结合的受体。
刺激性调节性G蛋白是与刺激性激素受体(Rs)相连的G蛋白,其α亚单位在激活时可以刺激酶或其他细胞内代谢的活性。相反,抑制性调节性G蛋白与抑制性激素受体相连,其α亚单位在激活时可以抑制酶或其他细胞内代谢的活性。
腺苷酸环化酶是一种12跨膜糖蛋白,它在Mg2+或Mn2+的辅助下催化ATP转化为cAMP。产生的cAMP是细胞代谢中的第二信使,是蛋白激酶A的变构激活剂。
蛋白激酶A是细胞代谢中的重要酶,因为它能够通过磷酸化特定的关键酶来调节细胞代谢。它还可以调节特定基因的表达、细胞分泌和膜通透性。这种蛋白酶包含两个催化亚单位和两个调节亚单位。在没有cAMP的情况下,复合物是不活跃的。当cAMP结合到调节亚单位时,它们的构象发生改变,导致调节亚单位的解离,激活蛋白激酶A并允许进一步的生物效应。
这些信号随后可以被cAMP磷酸二酯酶终止,这是一种将cAMP降解为5'-AMP并使蛋白激酶A失活的酶。
磷脂酰肌醇信号途径
在磷脂酰肌醇信号途径中,细胞外信号分子与细胞表面的G蛋白受体(Gq)结合并激活位于等离子膜上的磷脂酶C。这种脂酶水解磷脂酰肌醇4,5-二磷酸(PIP2)产生两个第二信使:肌醇1,4,5-三磷酸(IP3)和二酰甘油(DAG)。IP3与平滑内质网和线粒体膜上的IP3受体结合,打开Ca2+通道。DAG帮助激活蛋白激酶C(PKC),它通过磷酸化许多其他蛋白质,改变它们的催化活性,从而导致细胞反应。
Ca2+的作用也非常显著:它与DAG一起激活PKC,还可以激活CaM激酶途径,在该途径中,调节蛋白钙调蛋白(CaM)结合Ca2+,发生构象变化,并激活CaM激酶II,后者具有通过自磷酸化增加其与CaM的结合亲和力的独特能力,使CaM无法用于激活其他酶。然后激酶磷酸化靶酶,调节它们的活性。这两个信号途径通过Ca2+-CaM相互连接在一起,Ca2+-CaM也是cAMP信号途径中腺苷酸环化酶和磷酸二酯酶的调节亚单位。
受体调节
当GPCR长时间暴露于其配体时会变得不敏感。有两种公认的脱敏形式:1) 同源性脱敏,其中被激活的GPCR被降调;和2) 异源性脱敏,其中被激活的GPCR导致不同GPCR的降调。这种降调的关键反应是蛋白激酶对受体内部(或细胞质)区域的磷酸化。
cAMP依赖性蛋白激酶的磷酸化
环磷酸腺苷依赖性蛋白激酶(蛋白激酶A)由来自G蛋白(由受体激活)通过腺苷酸环化酶和环磷酸腺苷(cAMP)的信号链激活。在一种反馈机制中,这些激活的激酶磷酸化受体。受体保持活跃的时间越长,激活的激酶越多,磷酸化的受体就越多。在β2-肾上腺素受体中,这种磷酸化导致从Gs类G蛋白的耦合转换为Gi类。[43] cAMP依赖的PKA介导的磷酸化可以导致其他受体异源性脱敏。[44]
GRKs的磷酸化
G蛋白偶联受体激酶(GRKs)是只磷酸化活跃GPCR的蛋白激酶。[45] G蛋白偶联受体激酶(GRKs)是G蛋白偶联受体(GPCR)信号调节的关键调节器。它们构成了七种哺乳动物丝氨酸-苏氨酸蛋白激酶家族,这些激酶磷酸化结合激动剂的受体。GRKs介导的受体磷酸化迅速启动受体信号的显著损伤和脱敏。GRKs的活性和亚细胞定位通过与受体结构域、G蛋白亚单位、脂质、锚定蛋白和钙敏感蛋白的相互作用严格调节。[46]
受体的磷酸化可能有两个后果:
转位: 受体连同它所嵌入的膜部分一起被带入细胞内部,在那里在酸性囊泡环境中去磷酸化[47] 然后带回。这种机制用于调节长期暴露于某种激素的效应,例如通过允许脱敏后再敏感化。或者,受体可能经历溶酶体降解,或保持内化,在那里它被认为参与启动信号事件,其性质取决于内化囊泡的亚细胞定位。[44]
通过PKC和PKA对不同的丝氨酸/苏氨酸位点(以及IL-3和C-末端尾部)的磷酸化,可以以配体占据和GRK独立的方式增加对β-阿雷斯丁的亲和力。这些磷酸化通常足以自行损害G蛋白耦合。[48] PKC/PKA可能改为磷酸化GRKs,这也可能导致GPCR的磷酸化和β-阿雷斯丁的结合,以一种配体占据独立的方式进行。这两种后两种机制允许由于其他GPCR的活动而对一个GPCR进行脱敏,或称为异源性脱敏。GRKs也可能具有GAP结构域,因此可能通过非激酶机制促进失活。这些机制的组合也可能发生。
一旦β-阿雷斯丁与GPCR结合,它就会发生构象变化,允许其作为AP-2适配器复合体的支架蛋白,该复合体反过来召集另一种叫做clathrin的蛋白。如果足够多的受体在局部区域以这种方式招募clathrin,它们会聚集起来,由于clathrin分子之间的相互作用,膜向内突出,这个过程称为opsonization。一旦该凹陷从质膜上被amphiphysin和dynamin两种蛋白的作用剪切下来,它现在就成为一个内吞作用的泡。在这一点上,适配器分子和clathrin已经解离,受体要么被运输回质膜,要么被定位到溶酶体进行降解。
在这个过程的任何一个阶段,β-阿雷斯丁也可能招募其他蛋白——例如非受体酪氨酸激酶(nRTK),c-SRC——这些蛋白可能通过例如磷酸化小GTP酶,Ras,或直接招募ERK级联的蛋白(即Raf-1,MEK,ERK-1/2)来激活ERK1/2或其他有丝分裂原激活蛋白激酶(MAPK)信号,这时由于它们彼此之间的接近而启动信号。c-SRC的另一个靶标是参与内吞作用的dynamin分子。Dynamins围绕来临囊泡的颈部聚合,它们被c-SRC磷酸化提供了进行构象变化所需的能量,从而使最终的“从膜上剪切”成为可能。
GPCR细胞调节
受体脱敏是通过上述的磷酸化、β-阿雷斯丁结合和内吞作用的组合介导的。当内吞的受体嵌入一个被运输以与叫做溶酶体的细胞器融合的内体时,就会发生降调。因为溶酶体膜富含质子泵,它们的内部pH值较低(约4.8与pH约7.2的细胞质相比),这起到了变性GPCRs的作用。此外,溶酶体包含许多降解酶,包括蛋白酶,这些酶只能在如此低的pH下发挥作用,因此连接GPCR残基的肽键可能被切割。特定受体是否被运输到溶酶体、被扣留在内体中,或被运输回质膜取决于多种因素,包括受体类型和信号的大小。 GPCR调节还通过基因转录因子介导。这些因子可以增加或减少基因转录,从而增加或减少新受体的生成(上调或下调),这些受体运输到细胞膜。
受体寡聚化
G蛋白偶联受体(GPCR)寡聚化是一种广泛存在的现象。最为研究透彻的例子之一是代谢型GABAB受体。这种所谓的组成型受体是由GABABR1和GABABR2亚单位异源二聚化形成的。在异源系统中表达GABABR1而不表达GABABR2会导致该亚单位在内质网中滞留。而单独表达GABABR2亚单位则会导致该亚单位表面表达,尽管没有功能活性(即受体不结合激动剂,并且在暴露于激动剂后无法启动反应)。两个亚单位共同表达则导致功能性受体在质膜上表达。已经显示,GABABR2与GABABR1的结合掩盖了一个滞留信号[49],从而形成功能性受体。[50]
GPCR超家族的起源和多样化
由GPCR超家族介导的信号转导可以追溯到多细胞的起源。类似哺乳动物的GPCR在真菌中被发现,并根据基于GPCR指纹的GRAFS分类系统进行分类。[51] 在真核生物领域中对超家族成员的鉴定,以及对家族特定基序的比较,表明GPCR超家族具有共同的起源。[52] 特征基序表明,五个GRAFS家族中的三个,视紫红质、黏附和Frizzled,是在后鞭毛动物分裂之前从Dictyostelium discoideum cAMP受体演化而来的。后来,在线虫分裂之前,分泌素家族从黏附GPCR受体家族演化而来。[51] 昆虫GPCR似乎是自成一组,Taste2被识别为源自视紫红质。[52] 需要注意的是,分泌素/黏附的分裂是基于假定的功能而不是特征的,因为在研究中用经典的B类(7tm_2,Pfam PF00002)来识别两者。
参考资料
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. . Science. 2007, 318 (5854): 1258–65. PMC 2583103
 . PMID 17962520. doi:10.1126/science.1150577.
. PMID 17962520. doi:10.1126/science.1150577. - Filmore, David. . Modern Drug Discovery (American Chemical Society). 2004, 2004 (November): 24–28 [2012-03-29]. (原始内容存档于2018-09-08).
- Overington JP, Al-Lazikani B, Hopkins AL. . Nat Rev Drug Discov. December 2006, 5 (12): 993–6. PMID 17139284. doi:10.1038/nrd2199.
- Wettschureck N, Offermanns S. . Physiol. Rev. October 2005, 85 (4): 1159–204 [2016-10-18]. PMID 16183910. doi:10.1152/physrev.00003.2005. (原始内容存档于2017-12-03).
- Gilman A.G. . Annual Review of Biochemistry. 1987, 56: 615–649. PMID 3113327. doi:10.1146/annurev.bi.56.070187.003151.
- Royal Swedish Academy of Sciences. . 10 October 2012 [10 October 2012].
- Lindsley CW. . ACS Chemical Neuroscience. June 2013, 4 (6): 905–7. PMC 3689196 . PMID 24024784. doi:10.1021/cn400107y.
- Bjarnadóttir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schiöth HB. . Genomics. September 2006, 88 (3): 263–73. PMID 16753280. doi:10.1016/j.ygeno.2006.04.001.
- Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE; et al. . Proc Natl Acad Sci USA. 2003, 100 (8): 4903–4908 [2012-04-19]. PMC 153653 . PMID 12679517. doi:10.1073/pnas.0230374100. (原始内容存档于2020-08-28).
- Attwood TK, Findlay JB. . Protein Eng. 1994, 7 (2): 195–203 [2012-04-19]. PMID 8170923. doi:10.1093/protein/7.2.195. (原始内容存档于2007-10-12).
- Kolakowski LF Jr. . Receptors Channels. 1994, 2 (1): 1–7. PMID 8081729.
- Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, Spedding M, Harmar AJ. . Pharmacol Rev. 2005, 57 (2): 279–88. PMID 15914470. doi:10.1124/pr.57.2.5.
- . [2012-04-19]. (原始内容存档于2008-02-21).
- Joost P, Methner A. . Genome Biol. 2002, 3 (11): research0063.1–0063.16. PMC 133447 . PMID 12429062. doi:10.1186/gb-2002-3-11-research0063.
- Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB. . Genomics. 2006, 88 (3): 263–73. PMID 16753280. doi:10.1016/j.ygeno.2006.04.001.
- Xiao X, Wang P, Chou KC. . Journal of Computational Chemistry. 2009, 30 (9): 1414–1423. PMID 19037861. doi:10.1002/jcc.21163.
- Qiu JD, Huang JH, Liang RP, Lu XQ. . Anal. Biochem. July 2009, 390 (1): 68–73. PMID 19364489. doi:10.1016/j.ab.2009.04.009.
- Gu Q, Ding YS, Zhang TL. . Protein Pept. Lett. May 2010, 17 (5): 559–67. PMID 19594431. doi:10.2174/092986610791112693.
- Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. . Nature. June 2003, 423 (6941): 762–9. PMID 12802337. doi:10.1038/nature01705.
- Grigorieff N, Ceska TA, Downing KH, Baldwin JM, Henderson R. . J. Mol. Biol. 1996, 259 (3): 393–421. PMID 8676377. doi:10.1006/jmbi.1996.0328.
- Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. . Nature. 1997, 389 (6647): 206–11. PMID 9296502. doi:10.1038/38323.
- Pebay-Peyroula E, Rummel G, Rosenbusch JP, Landau EM. . Science. 1997, 277 (5332): 1676–81. PMID 9287223. doi:10.1126/science.277.5332.1676.
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Trong IL, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. . Science. 2000, 289 (5480): 739–45. PMID 10926528. doi:10.1126/science.289.5480.739.
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. . Nature. 2007, 450 (7168): 383–7. PMID 17952055. doi:10.1038/nature06325.
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. . Science. 2007, 318 (5854): 1266–73. PMID 17962519. doi:10.1126/science.1150609.
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. . Nature. January 2011, 469 (7329): 175–80. PMC 3058308 . PMID 21228869. doi:10.1038/nature09648.
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. . Nature. January 2011, 469 (7329): 236–40. PMC 3074335 . PMID 21228876. doi:10.1038/nature09665.
- Warne T, Moukhametzianov R, Baker JG, Nehmé R, Edwards PC, Leslie AG, Schertler GF, Tate CG. . Nature. January 2011, 469 (7329): 241–4. PMC 3023143 . PMID 21228877. doi:10.1038/nature09746.
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. . Science. April 2011, 332 (6027): 322–7. PMC 3086811 . PMID 21393508. doi:10.1126/science.1202793.
- Rasmussen SG, Devree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. . Nature. July 2011, 477 (7366): 549–55. PMC 3184188 . PMID 21772288. doi:10.1038/nature10361.
- Stuart JA, Brige RR. . Lee AG (编). . Greenwich, Conn: JAI Press. 1996: 33–140. ISBN 1-55938-659-2.
- Hofmann KP, Heck M. . Lee AG (编). . Greenwich, Conn: JAI Press. 1996: 141–198. ISBN 1-55938-659-2.
- Kolb H, Fernandez E, Nelson R, Jones BW. . University of Utah. 2010-03-01. (原始内容存档于2000-08-16).
- Bakthavatsalam D, Brazill D, Gomer RH, Eichinger L, Rivero F, Noegel AA. . Curr Biol. 2007, 17 (10): 892–7. PMID 17481898. doi:10.1016/j.cub.2007.04.029.
- Hazell GG, Hindmarch CC, Pope GR, Roper JA, Lightman SL, Murphy D, O'Carroll AM, Lolait SJ. . Front Neuroendocrinol. July 2011, 33 (1): 45–66. PMC 3336209 . PMID 21802439. doi:10.1016/j.yfrne.2011.07.002.
- Dorsam RT, Gutkind JS. . Nature Reviews. Cancer. February 2007, 7 (2): 79–94. PMID 17251915. doi:10.1038/nrc2069.
- Lagerstrom MC; et al. . Nat Rev Drug Discov. April 2008, 7 (4): 339–57. PMID 18382464.
- Bhattacharya S; et al. . J Comput Aided Mol Des. August 2010, 24 (8): 659–74. PMID 20512399.
- Digby GJ, Lober RM, Sethi PR, Lambert NA. . Proc. Natl. Acad. Sci. U.S.A. November 2006, 103 (47): 17789–94. PMC 1693825 . PMID 17095603. doi:10.1073/pnas.0607116103.
- Smith JS, Nicholson LT, Suwanpradid J, Glenn RA, Knape NM, Alagesan P, et al. . Science Signaling. November 2018, 11 (555): eaaq1075. PMC 6329291 . PMID 30401786. doi:10.1126/scisignal.aaq1075.
- Kim JY, Haastert PV, Devreotes PN. . Chemistry & Biology. April 1996, 3 (4): 239–43. PMID 8807851. doi:10.1016/S1074-5521(96)90103-9 .
- Duchene J, Schanstra JP, Pecher C, Pizard A, Susini C, Esteve JP, et al. . The Journal of Biological Chemistry. October 2002, 277 (43): 40375–83. PMID 12177051. doi:10.1074/jbc.M202744200 .
- Chen-Izu Y, Xiao RP, Izu LT, Cheng H, Kuschel M, Spurgeon H, Lakatta EG. . Biophysical Journal. November 2000, 79 (5): 2547–56. PMC 1301137 . PMID 11053129. doi:10.1016/S0006-3495(00)76495-2.
- Tan CM, Brady AE, Nickols HH, Wang Q, Limbird LE. . Annual Review of Pharmacology and Toxicology. 2004, 44 (1): 559–609. PMID 14744258. doi:10.1146/annurev.pharmtox.44.101802.121558.
- Santulli G, Trimarco B, Iaccarino G. . High Blood Pressure & Cardiovascular Prevention. March 2013, 20 (1): 5–12. PMID 23532739. S2CID 45674941. doi:10.1007/s40292-013-0001-8.
- Penela P, Ribas C, Mayor F. . Cellular Signalling. November 2003, 15 (11): 973–81. PMID 14499340. doi:10.1016/S0898-6568(03)00099-8.
- Krueger KM, Daaka Y, Pitcher JA, Lefkowitz RJ. . The Journal of Biological Chemistry. January 1997, 272 (1): 5–8. PMID 8995214. doi:10.1074/jbc.272.1.5 .
- Tobin AB. . British Journal of Pharmacology. March 2008, 153 (Suppl 1): S167–76. PMC 2268057 . PMID 18193069. doi:10.1038/sj.bjp.0707662.
- Margeta-Mitrovic M, Jan YN, Jan LY. . Neuron. July 2000, 27 (1): 97–106. PMID 10939334. S2CID 15430860. doi:10.1016/S0896-6273(00)00012-X .
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, et al. . Nature. December 1998, 396 (6712): 679–82. Bibcode:1998Natur.396..679W. PMID 9872316. S2CID 4406311. doi:10.1038/25354.
- Krishnan A, Almén MS, Fredriksson R, Schiöth HB. Xue C , 编. . PLOS ONE. 2012, 7 (1): e29817. Bibcode:2012PLoSO...729817K. PMC 3251606 . PMID 22238661. doi:10.1371/journal.pone.0029817 .
- Nordström KJ, Sällman Almén M, Edstam MM, Fredriksson R, Schiöth HB. . Molecular Biology and Evolution. September 2011, 28 (9): 2471–80. PMID 21402729. doi:10.1093/molbev/msr061.
外部链接
| 维基共享资源上的相关多媒体资源:G蛋白偶联受体 |
- 醫學主題詞表(MeSH):G-protein-coupled+receptors
- (英文)GPCR Cell Line
- (英文). IUPHAR Database. International Union of Basic and Clinical Pharmacology. [11 August 2008]. (原始内容存档于2012-03-20).
- (英文). [2018-03-31]. (原始内容存档于2020-10-27).
Data, diagrams and web tools for G protein-coupled receptors (GPCRs).
; Munk C, Isberg V, Mordalski S, Harpsøe K, Rataj K, Hauser AS, Kolb P, Bojarski AJ, Vriend G, Gloriam DE. . British Journal of Pharmacology. 2016, 173 (14): 2195–207. PMC 4919580. PMID 27155948. doi:10.1111/bph.13509.